
Актуальність питання
Існує велика потреба у розробці високоточних економічно ефективних технологій, які б сприяли широкому практичному впровадженню неінвазивного пренатального тестування (НІПТ).
Методи
Ми розробили метод, що базується на прицільному (таргетному) аналізі позаклітинної ДНК для детекції фетальних анеуплоїдій хромосом 13, 18 та 21. Цей метод дозволяє захоплювати та аналізувати певні обрані регіони інтересу в геномі. Аналітичні характеристики методу вивчались у рамках сліпого дослідження на 631 зразках, взятих у жінок з терміном гестації принаймні 10 тижнів, яким також виконували інвазивне тестування.
Результати
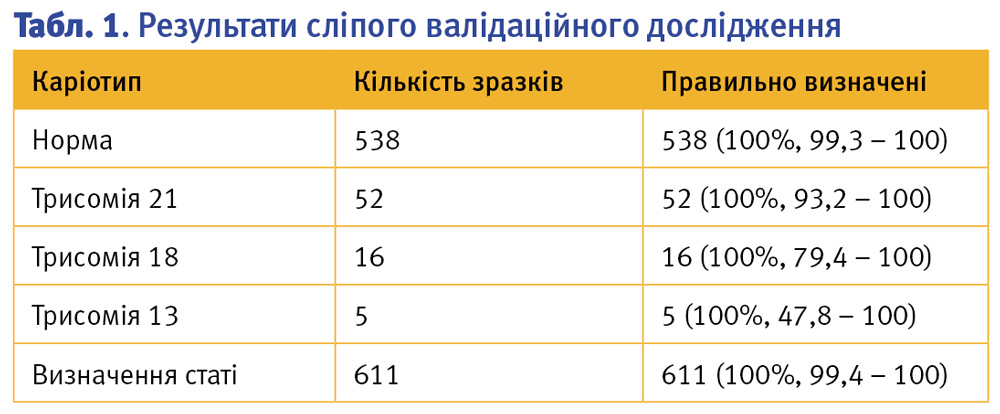
Наше сліпе дослідження показало діагностичну чутливість та специфічність 100% та правильно визначило 52/52 (95% ДІ, 93,2% – 100%) випадків трисомії 21, 16/16 (95% ДІ, 79,4% – 100%) випадків трисомії 18, 5/5 (95% ДІ, 47,8% – 100%) випадків трисомії 13 та 538/538 (95% ДІ, 99,3% – 100%) випадків без патології. Цей тест також правильно визначив стать плода в усіх випадках (95% ДІ, 99,4% – 100%). Один зразок не відповідав заздалегідь визначеним для цього методу критеріям контролю якості, і ще по 19 зразках результати не видавали у зв’язку із низькою фетальною фракцією.
Висновки
Обсяги впровадження тесту фетальної ДНК як методу універсального скринінгу на трисомії 13, 18 та 21 залежать в основному від його точності та вартості. Аналіз таргетних регіонів геному позаклітинної ДНК у материнській плазмі крові забезпечує точну неінвазивну детекцію фетальних анеуплоїдій та є економічно вигідним методом, а це найважливіші передумови для широкого впровадження НІПТ у практику.
Вступ
Відкриття вільної фетальної ДНК (вфДНК) у крові матері [1] ознаменувало собою початок ери неінвазивного пренатального тестування (НІПТ) та призвело до розробки перших неінвазивних тестів. Вільна фетальна ДНК у материнській плазмі крові успішно застосовувалась для визначення статі плода та наявності гемолітичної хвороби у плода [2, 3]. У низці клінічних лабораторій у різних країнах світу ці методи вже стали стандартними. Однак прямий аналіз невеликої кількості вфДНК у присутності великої кількості власної материнської ДНК становить велику проблему для НІПТ.
Раніше вважалось, що частка вфДНК у крові матері становить близько 3–6% від загальної кількості позаклітинної ДНК [4]. Однак, останні дослідження вважають, що фетальна фракція може складати навіть 10–20% [5]. Присутність великої кількості материнської ДНК у крові матері порівняно із невеликою кількістю фетальної ДНК становить велику проблему для кількісного визначення фетальної ДНК і для детекції фетальних анеуплоїдій.
За останнє десятиліття застосовували велику кількість різноманітних методів для того, щоб розрізнити вфДНК та материнську ДНК або збагатити вфДНК для її кращої ідентифікації [6].
До таких методів роботи з ДНК відносяться секвенування та епігенетичні методи, які фокусують свою увагу на дослідженні метиляції фетальної ДНК за допомогою обробки ДНК натрію бісульфатом [7], застосування рестрикційної ендонуклеази, чутливої до метиляції, та застосування антитіл, специфічних до 5-метил-цитозинових залишків динуклеотидів CpG у геномі [8–10]. Застосовувались і альтернативні методи вивчення фетально-специфічної мРНК [11] та дослідження фетально-специфічних білків [12].
Застосування технологій секвенування нового покоління (NGS) у сфері НІПТ стало справжньою революцією у цій сфері. У 2008 р. дві незалежні групи вчених продемонстрували, що НІПТ для трисомії 21 (Т21) можна провести, застосувавши масивне паралельне секвенування методом shotgun (метод «дробовика») [13, 14], що відкрило нову еру НІПТ та нові можливості для застосування цих технологій у клінічній практиці. На основі цих результатів біотехнологічні компанії та незалежні групи вчених започаткували клінічні дослідження та розробили нові тести НІПТ [15–21].
Нещодавно були розроблені прицільні (таргетні) методи NGS, які застосовують лише певні специфічні послідовності, що cтановлять інтерес. Описані та представлені методи NGS, що базуються на визначенні поліморфізму одного нуклеотиду із застосуванням мультиплексної таргетної ампліфікації для аналізу цих окремих поліморфізмів; а також кількісний метод NGS, який застосовує ліговані зонди з їх подальшою ампліфікацією та секвенуванням [20, 21]. Таргетні методи мають потенціал збільшити пропускну спроможність та зменшити витрати, тому що для них потрібен значно менший обсяг секвенування, ніж для методів секвенування повного геному.
Попри все це, однак, існує велика потреба у розробці ще точніших та економічно доцільних методів НІПТ. Зокрема, надзвичайно корисними видаються методи, які націлені на певні специфічні послідовності, що становлять інтерес, це означає, що секвенування знадобиться лише у певному потрібному обсязі на відміну від секвенування повного геному. Тут ми представляємо високоточний та економічно доцільний метод детекції фетальних трисомій 13, 18, та 21, який долає багато недоліків, притаманних поточним технологіям НІПТ.
Матеріали та методи
Забір зразка
Були отримані зразки плазми крові вагітних жінок 18 років та старше з терміном гестації принаймні 10 тижнів. Персональні дані не зазначались. Проводили аналіз лише одноплодових вагітностей. Протоколи забору зразків були ухвалені Національними комітетами з біоетики, від усіх учасниць отримана поінформована згода. Клініки, які скеровували пацієнток на дослідження, отримали усю відповідну інформацію щодо критеріїв включення в це дослідження, його переваг та недоліків [22]. Типами анеуплоїдій, включеними в це дослідження, були трисомії 21, 18 та 13, що було підтверджено інвазивним тестуванням.
Підготовка зразків
У кожної з учасниць дослідження взяли в середньому по 8 мл периферичної крові в пробірки з EDTA. Було отримано в середньому 4 мл плазми крові за допомогою подвійного центрифугування за протоколом 1600 х g впродовж 10 хв. з подальшим режимом 16 000 х g впродовж 10 хв. Зразки плазми крові отримали унікальний ідентифікатор, їх помістили на зберігання за температури –80°С до моменту їх аналізу. Із 4 мл плазми крові за допомогою тест-системи Qiasymphony DSP Virus/Pathogen Midi Kit (Qiagen) екстрагували вфДНК.
Підготовка бібліотеки секвенування
Обробка екстрагованої ДНК проводилася за допомогою стандартних методів підготовки лабораторії з незначними модифікаціями [23]. Також були підготовані бібліотеки контролів з негативними результатами. В цілому, «липкі» кінці 5’ і 3’ були «добудовані» за допомогою Т4 полімерази (NEB), та 5’ фосфати були приєднані за допомогою Т4 полінуклеотидкінази (NEB). Продукти реакції були очищені за допомогою тест-системи MinElute (Qiagen). Після цього адаптери секвенування були ліговані з обома кінцями ДНК за допомогою Т4 ДНК-лігази (NEB), після чого виконували очищення за допомогою тест-системи MinElute (Qiagen). Одноланцюгові розриви прибрали в реакції «добудови» за допомогою Bst-полімерази (NEB) з подальшою інкубацією при 65°С впродовж 25 хв., а потім при 12°С впродовж 20 хв. Ампліфікацію бібліотеки виконано за допомогою полімерази Fusion (Agilent Technologies), усім зразкам присвоєний свій унікальний штрих-код. Продукти секвенування бібліотеки було очищено за допомогою тест-системи очищення MinElute (Qiagen).
Дизайн та підготовка таргетних послідовностей
Були розроблені спеціальні таргетні послідовності (TACS) розміром приблизно 250 пн з метою захоплення певних обраних локусів хромосом 21, 18, 13 та Y (див. Табл. 1 у Додатку до онлайнової версії цієї статті за посиланням www.clinchem.org/content/vol62/issue6). Таргетні локуси геному були вибрані на основі контенту (вмісту) GC, відстані від повторюваних елементів та відсутності поруч складної архітектури геному. TACS були підготовані полімеразною ланцюговою реакцією за допомогою MyTaq-полімерази (Bioline) та праймерів, розроблених для ампліфікації таргетних локусів нормальної ДНК. Амплікони верифіковані електрофорезом в агарозному гелі та очищені стандартними системами ПЛР-очищення, такими як Qiaquick (Qiagen) та NucleoSpin 96 (Macherey Nagel). Концентрацію TACS вимірювали спектрофотометром Nanodrop (Thermo Scientific).
Був створений еквімолярний пул TACS з тупими кінцями, тупі кінці виконано за допомогою тест-системи Quick Blunting (NEB). Після очищення за допомогою тест-системи MinElute (Qiagen) їх було біотинільовано за допомогою тест-системи Quick Ligation (NEB) та очищено за допомогою тест-системи MinElute (Qiagen). Після цього TACS (1500 нг) імобілізували на магнітних часточках, вкритих стрептавідином (Invitrogen), як зазначалось вище [24].
Гібридизація
Ампліфіковані бібліотеки змішали з буфером для гібридизації (Agilent), блокуючим агентом (Agilent), блокуючими олігонуклеотидами [25], Cot-1 ДНК (Invitrogen) та ДНК сперми лосося (Invitrogen). Потім гібридизаційні суміші бібліотеки секвенування денатурували при 95°С впродовж 3 хв. та інкубували при 37°С впродовж 20 хв., перед тим, як додати до біотинільованих TACS. Після цього зразки інкубували 12–48 год. при 66°С та промивали, як зазначено вище [24]. Захоплені послідовності піддавали елюації шляхом нагрівання. Елюйовані послідовності були ампліфіковані за допомогою зовнішніх адаптерних праймерів. Був створений еквімолярний пул збагачених продуктів ампліфікації, секвенування виконували на платформі секвенування MiSeq, NextSeq 500 або Hiseq 2500 (Illumina).
Аналіз даних
Вирівнювання до референтного геному людини. Спарені кінцеві фрагменти кожного із зразків обробляли за допомогою програмного забезпечення Cutadapt [26] з метою видалення адаптерних послідовностей та результатів поганої якості. Послідовності, що залишилися, вирівняли до структури референтного геному людини hg19 (USCS Genome Bioinformatics) за допомогою алгоритму вирівнювання Барроуз–Уіллер [27]. Для того, щоб прибрати дублікати зчитуваних фрагментів та перетворити вирівняні фрагменти в бінарний файл (ВАМ), в якому містяться унікальні вирівняні зчитані фрагменти, застосували інструментарій програмного забезпечення Picard [Broad Institute (2015) Picard]. Із цього фінального файлу ВАМ була отримана інформація про глибину зчитування по парам нуклеотидів за допомогою програмного забезпечення SAMtools. Інформація про однонуклеотидні поліморфізми в таргетних послідовностях була отримана за допомогою програмного пакету функцій bcftools та сценарію виконання vcfutils.pl, що додаються до пакету програмного забезпечення SAMtools [27].
Класифікація фетальних анеуплоїдій. Процедура секвенування виявляє розбіжність глибини зчитування в багатьох регіонах інтересу. Це частково залежить від вмісту GC кожного з секвенованих регіонів [28]. Зменшення GC-зсуву було досягнуто шляхом визначення вмісту GC кожного з регіонів і подальшого групування схожих за глибиною зчитування GC-вмісних регіонів для створення погоджених груп за принципом схожості. Погоджені групи досліджуваної хромосоми порівнювали з відповідними погодженими групами референтної хромосоми за допомогою 3 статистичних критеріїв: парного t-критерію, двомірної непараметричної програми самозавантаження (a bivariate nonparametric bootstrap) та стратифікованого критерію перестановок. Для обчислення зваженої суми використовували показники, отримані при застосуванні кожного з методів. Для врахування систематичної похибки від постановки до постановки [29] кожну зважену суму було нормалізовано шляхом віднімання медіани, специфічної для постановки, та ділення на кратну суму емпіричного стандартного відхилення еуплоїдних зразків. Медіану, специфічну для постановки, обчислювали із зважених сум усіх зразків однієї постановки секвенування. Теоретична дисперсія випадкової величини, яку відображає зважена сума усіх 3 методів, була обчислена з набору 100 еуплоїдних зразків. Цей нормалізований показник використали для обчислення ризику трисомії (tririsk) для кожного зразка. Показники, які були вищими певного порогового значення, були віднесені до категорії високого ризику трисомії.
Визначення фетальної фракції. Методом байєсівського висновку [30] була розроблена кінцева модель суміші біноміальних розподілень, яку використали для обчислення апостеріорного розподілу фетальної фракції ДНК шляхом підрахунку алелей гетерозиготних локусів у материнській плазмі крові. У цій моделі було використано три можливі інформативні комбінації генотипів матері/плода для кількісного визначення фетальної фракції ДНК, що повністю відповідало даним, що спостерігалися. Апостеріорний розподіл фетальної фракції обчислювали за допомогою алгоритму Метрополіса–Гастінгса [31]. У подальшому була логічно виведена нижня межа 95%-го довірчого інтервалу такого апостеріорного розподілу ймовірностей.

коментарів