

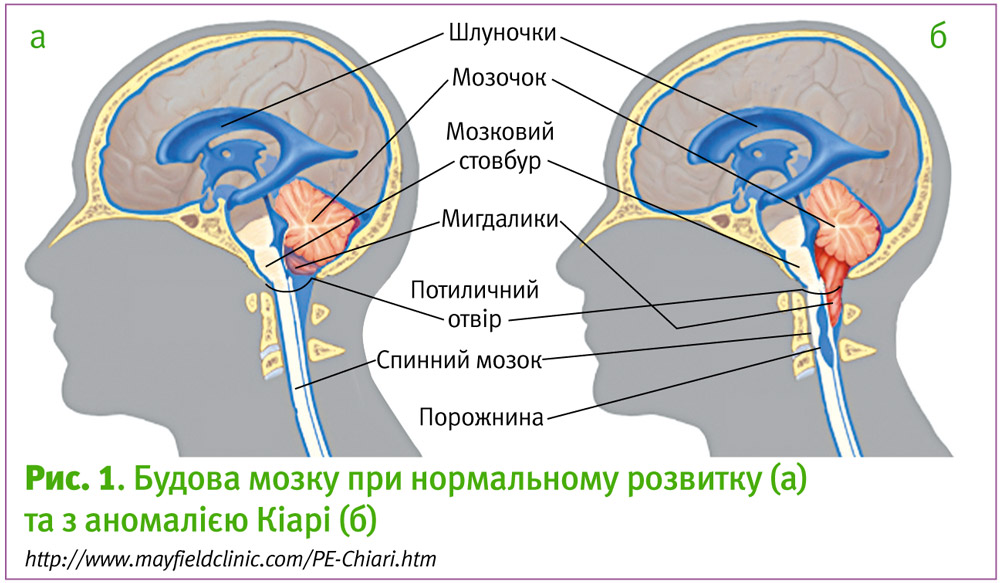
Аномалія Кіарі – група вроджених захворювань, при яких спостерігається зміщення донизу задньої частини мозочка або поєднання опущення мозочка та нижньої частини довгастого мозку через великий потиличний отвір (foramen magnum) у спинномозковий канал [7]. Така патологія часто супроводжується підвищенням лікворного та внутрішньочерепного тиску, що необхідно враховувати під час планування та проведення анестезії
У доступній англомовній літературі ми не знайшли чітких рекомендацій, заснованих на рандомізованих контрольованих дослідженнях, щодо вибору тактики розродження та проведення анестезії під час кесарського розтину у даної категорії пацієнток. У нашому пологовому будинку «Лелека» (Київ) трапилась така пацієнтка, тому ми хочемо поділитись клінічним випадком та обговорити цю патологію.
Вперше аномалія Кіарі була описана Cleland у 1883 році [7]. Етіологія та патогенез даного захворювання залишається предметом дискусії.
Запропоновано декілька теорій розвитку аномалії Кіарі [1].
- Молекулярно-генетична теорія передбачає, що патологія розвивається внаслідок первинного дефекту генів, що відповідають за сегментацію заднього мозку та розвиток потиличної кістки і відповідних анатомічних структур [2]. Враховуючи те, що більшість випадків аномалії Арнольда-Кіарі зустрічаються спорадично і не є набутими, причинами дефекту генів є, очевидно, спонтанна мутація чи делеція гена, або мутація, спричинена впливом певного тератогена.
- Гідродинамічна теорія. Згідно з цією теорією, опущення мозочка та стовбура мозку спричинено ранньою прогресуючою гідроцефалією плода [3].
- Компресійна теорія припускає обмежений ріст задньої черепної ямки, у зв’язку з чим виникає невідповідність її розмірів розмірам мозочка. Це викликає компресію нервової тканини і протискання її у великий потиличний отвір [4, 5].
- Теорія недостатності спинномозкової рідини. Дефект закриття нервової трубки на ранніх термінах розвитку плода викликає зниження об’єму спинномозкової рідини, і його недостатньо для повноцінного розтягнення шлуночкової системи, що веде до зменшення розмірів задньої черепної ямки [6].
Є чотири анатомічні варіанти аномалії Кіарі, які виділив Chiari у 1891 році [8, 12].
І тип характеризується аномальною формою мигдалин мозочка, які зміщені нижче рівня великого потиличного отвору. Гідроцефалія при цьому варіанті зустрічається рідко. Зазвичай, І тип аномалії Арнольда-Кіарі зумовлений порушенням розвитку мезодерми, але також описані нейроектодермальні та набуті форми [8, 12].
ІІ тип зустрічається найчастіше. Він також називається аномалією Арнольда-Кіарі. Спостерігається опущення нижньої частини черв’яка (vermis) та півкуль мозочка через великий потиличний отвір з дислокацією стовбуру мозку (середній мозок, четвертий шлуночок і нижня частина моста). Цей варіант асоціюється зі spina bifida та іншими аномаліями розвитку головного мозку, спинного мозку та мозкових оболонок. Гідроцефалія присутня у 70% випадків і носить констриктивний або обструктивний характер [9, 12].
ІІІ тип зустрічається рідко і поєднує у собі малу задню черепну ямку з високою шийною або потиличною енцефальною грижею, у яку зазвичай зміщуються і структури мозочка. Також часто спостерігається зміщення стовбура мозку донизу в спинномозковий канал. Гідроцефалія спостерігається у 50% випадків і має обструктивний характер внаслідок стенозу водогону мозку або наявності мальформації Dandy-Walker [9, 12].
IV тип у наші дні вважається застарілим терміном, який описує гіпоплазію мозочка, що не пов’язана з іншими типами аномалії Кіарі.
На додаток до класичних чотирьох типів аномалії Кіарі виділяють додаткові підтипи [8, 12]. Кіарі 0 характеризується анатомічною деформацією стовбура мозку (задній нахил моста, заднє зміщення середнього мозку, низьке розташування перепони (obex) при нормальному розташуванні мигдалин мозочка [10]. Кіарі 1,5 подібна до Кіарі II, але без spina bifida [11]. Кіарі 3,5 поєднує у собі Кіарі III і патологічне сполучення між нервовою трубкою і травним трактом (foregut) [12]. Аномалія Кіарі зустрічається у 0,1–0,5% населення [13].
Клінічні прояви
Аномалія Кіарі проявляється широким спектром симптомів, обумовлених складним патогенезом, і вони залежать від конкретного варіанта патології. При Кіарі I можуть розвиватися охриплість, параліч голосових зв’язок, дизартрія, дисфункція м’якого піднебіння, фарингеальна ахалазія, атрофія язика, аспірація, ністагм, центральне та обструктивне апное уві сні [1, 8, 14, 15], осцилопсія, сенсоневральна глухота, синусова брадикардія, синкопе, гикавка, загальна слабкість, гіперрефлексія, позитивний симптом Бабінського, скандована мова, атаксія [16], головний біль, сколіоз [17], а також сенсорний і моторний неврологічний дефіцит, спричинений сирінгомієлією.
При Кіарі II можуть спостерігатися дисфагія, слабкість у руках, стридор, періодичне апное, аспірація, сколіоз [18], ознаки оклюзивної гідроцефалії та сирінгомієлії.
Хворі з аномалією Кіарі III типу у більшості випадків помирають у період новонародженості внаслідок дихальних розладів. У тих, хто вижив, розвиваються важкі неврологічні розлади – затримка розумового розвитку, епілепсія, гіпотонія або спастичність, парези та паралічі верхніх та нижніх кінцівок, параліч нижніх черепних нервів.
Лікування
Тактика лікування аномалії залежить від її типу та особливостей патологічної анатомії. I тип за відсутності симптомів та сирінгомієлії може спостерігатися консервативно з проведенням повторного МРТ через 6 місяців і далі – один раз на рік [19]. За наявності таких ознак, як параліч черепних нервів, сирінгомієлія, мієлопатія, мозочкова симптоматика, біль у шиї чи потилиці, показане хірургічне лікування [20]. Воно полягає у декомпресії краніоцервікального з’єднання та відновленні нормальної циркуляції спинномозкової рідини у ділянці великого потиличного отвору. До методик оперативного лікування належать задня декомпресія великого потиличного отвору (з відкриттям твердої мозкової оболонки або без) і передня трансоральна одонтоїдектомія [20]. Хірургічне лікування II та III типу аномалії Кіарі може включати закриття дефектів нервової трубки одразу після народження, лікування гідроцефалії шляхом шунтування та декомпресію задньої черепної ямки [1].
Вплив вагітності на перебіг аномалії Арнольда-Кіарі
У більшості випадків розродження проводиться шляхом кесарського розтину. Прийом Вальсальви у першому періоді пологів може призвести до значного погіршення неврологічної симптоматики. За відсутності клінічних проявів, а також у вагітних, яким проводилася хірургічна декомпресія, пологи можна провести консервативно з виключенням потужного періоду (що практично не використовується у сучасній практиці) [24].
Існують певні особливості анестезіологічного забезпечення при кесарському розтині. У пацієнток із проведеною хірургічною декомпресією, а також без неї, застосовується як загальна, так і епідуральна анестезія [21, 22]. Якщо є ознаки підвищеного внутрішньочерепного тиску і хірургічна декомпресія не проводилася, анестезією вибору є загальна анестезія [22].
При виконанні загальної анестезії потрібно уникати поверхневого рівня анестезії, застосовуючи наркотичні анальгетики до вилучення плоду та обов’язково поставивши до відома неонатологів. У літературі описаний летальний випадок після знеболення пологів методом епідуральної аналгезії та ненавмисної перфорації твердої мозкової оболонки (наслідок вклинення довгастого мозку у великий потиличний отвір) у пацієнтки з внутрішньочерепною гіпертензією [23]. За результатами розтину у хворої виявлено недіагностовану пухлину головного мозку. Подібна ситуація, теоретично, може виникнути і у пацієнта з аномалією Кіарі. Тому перед анестезіологом постає питання у виборі методу знеболення як пологів, так і кесаревого розтину.
Клінічний випадок
Жінка, 29 років, вперше народжує, ризик за Американською асоціацією анестезіологів (ASA) – 1, надійшла у клініку в плановому порядку на 39 тижні вагітності.
Неврологічний анамнез ускладнений фокальною епілепсією. Перший напад епілепсії, зі слів хворої, був зареєстрований у 2015 році. Від специфічного протиепілептичного лікування відмовилась. При обстеженні у зв’язку з епілепсією було проведено магнітно-резонансну томографію (МРТ) головного мозку, при якій виявлено мальформацію Арнольда-Кіарі І типу. При консультації з нейрохірургом докладне обстеження не виявило неврологічного дефіциту та специфічних симптомів захворювання.
Було рекомендоване консервативне ведення захворювання з проведенням контрольної МРТ головного мозку один раз на рік. Нейрохірургом не було виявлено протипоказів до кесарського розтину, загальної та нейроаксіальної анестезії.
Супутні захворювання пацієнтки включали гіпохромну анемію легкого ступеня (Hb=9,7 г/дл), хронічний цистит у стадії ремісії, хронічний холецистит у стадії ремісії, виразкову хворобу шлунка в стадії ремісії, міопію легкого ступеня. У віці 19 років хворій була проведена кріодеструкція з приводу ерозії шийки матки. Враховуючи тазове передлежання плода, відмову жінки від спроби вагінальних пологів, а також те, що під час пологів при виконанні прийому Вальсальви можливе значне підвищення внутрішньочерепного тиску, що може провокувати розвиток неврологічної симптоматики, пов’язаної з синдромом Кіарі, було прийнято рішення про проведення планового (елективного) кесарського розтину. Передопераційне обстеження включало фізикальне обстеження та лабораторне обстеження (загальний аналіз крові, загальний аналіз сечі, коагулограма), консультацію нейрохірурга. Маса тіла пацієнтки до вагітності становила 48,5 кг, на момент прийому – 64,3 кг. Зріст – 160 см. Дані фізикального обстеження без особливостей, артеріальний тиск – 122/78 мм рт. ст., пульс – 78 уд./хв., частота дихання – 15/хв., Sp02 – 98%. За результатами акушерського огляду виявлено один живий плід, поздовжнє положення, тазове передлежання. ЧСС плода – 140/хв., передбачувана вага – 3958 г. Шийка матки вкорочена до 1,5 см та розкрита на 1 см, 3 бали за шкалою Бішопа.
За результатами лабораторного обстеження, окрім анемії, патології не виявлено. На консультації у нейрохірурга встановлено, що протипоказів до кесарського розтину та проведення анестезії немає, можлива як загальна, так і регіонарна анестезія.
У день операції хворій катетеризовано периферичну вену, за одну годину до розрізу шкіри введено внутрішньовенно 1 грам ампіциліну. Після подачі в операційну у хворої оцінено артеріальний тиск, пульс, сатурацію периферичної крові та розпочато швидку інфузію збалансованого кристалоїдного розчину.
Одночасно з початком інфузії в положенні сидячи, в асептичних умовах голкою Тухі 18G на рівні L3-L4 під місцевим знеболенням шкіри 1% розчином лідокаїну 4,0 мл проведено пункцію епідурального простору. Ідентифікація епідурального простору проводилася за допомогою шприца низького опору методом втрати опору. Введено епідуральний катетер на 3 сантиметри краніально. Катетер фіксовано до шкіри, накладено асептичну пов’язку. Після аспіраційної проби введено тест-дозу ропівакаїну. Пацієнтку переведено у положення лежачи, налагоджено моніторинг АТ, ЕКГ, ЧСС, Sp02, ЧД. З метою профілактики аортокавальної професії операційний стіл нахилено вліво на 15о. Через 5 хв. після введення тест-дози ознак спінального блоку не виявлено. Неінвазивне вимірювання артеріального тиску проводилося кожні дві хвилини. Тиск при надходженні в операційну – 127/82 мм Hg, пульс – 84/хв. Введено епідурально 20 мл 0,75% ропівакаїну. Через 18 хв. після введення отримано повний сенсомоторний блок на рівні Т6. Проведено катетеризацію сечового міхура.
На момент початку операції АТ – 108/72 мм рт. ст., пульс – 82/хв. З метою профілактики гіпотензії розпочато інфузію фенілефрину зі швидкістю 0,5 мг/годину та продовжено швидку інфузію збалансованого кристалоїдного розчину. Розпочато кесарський розтин за Штарком із доступом по Джо-Когану. Вилучення дитини відбулося на першій хвилині, пуповина пересічена після закінчення пульсації. Отримано одну живу дитину масою 3550 г та довжиною тіла 53 см. Після пересічення пуповини введено 5 Од окситоцину в/в. Оцінка за Апгар на першій хвилині – 9 балів, на п’ятій хвилині – 10 балів. Відділено плаценту, матка ушита однорядним безперервним швом. Об’єм інфузії під час операції становив 500 мл збалансованого кристалоїдного розчину. Тривалість операції – 32 хв.
Ранній післяопераційний період без особливостей. Через 4 години пацієнтка була мобілізована. Післяопераційне знеболення включало декскетопрофен 50 мг в/в кожні 8 годин та ропівакаїн 0,2% по 10 мл епідурально кожні три години протягом доби. Через 8 годин після операції пацієнтка була переведена у акушерське відділення. На третю добу після операції у пацієнтки відбувся судомний напад з втратою свідомості, який був знятий внутрішньовенним введенням діазепаму 10 мг. Пацієнтка повторно госпіталізована до відділення анестезіології та інтенсивної терапії, проведені консультація невролога та електроенцефалографія, на якій виявлено підвищену судомну активність. Також проведене повне лабораторне обстеження. Після консультації невропатолога був виставлений діагноз: Криптогенна епілепсія з простими та комплексними парціальними моторними нападами. Синдром Арнольда-Кіарі I. Рекомендовано лікування: 1) нормалізація режиму сну; 2) Прийом топіромату (топомакс, топіромакс) по 25 мг на ніч 1–2 доби, потім 25 мг двічі на день 1 тиждень, потім по 37,5 мг двічі на добу з динамічним спостереженням за хворою. Можливе збереження грудного вигодовування, враховуючи безпечний профіль препарату.
Дискусія
На нашу думку, використання епідуральної анестезії є безпечним у пацієнток із асимптомною формою аномалії Кіарі і може застосовуватися, навіть якщо не було проведено хірургічної корекції. Її переваги, якщо порівнювати з загальною анестезією, полягають у відсутності медикаментозного впливу на плід та можливістю первинного контакту дитини з матір’ю «шкіра до шкіри» ще в операційній, а також в адекватному післяопераційному знеболенні без використання опіоїдів. Але є потенційний ризик ненавмисної пункції твердої мозкової оболонки. Теоретично, якщо у пацієнтки є нерозпізнана внутрішньочерепна гіпертензія, то зниження лікворного тиску може спричинити дислокацію стовбура мозку через великий потиличний отвір і, як наслідок, призвести до фатальних наслідків. Тому ми вважаємо, що рішення про вибір методу анестезії повинне прийматися колегіально анестезіологом разом із нейрохірургом та акушером-гінекологом.
Розвиток судом на третій день після операції, на нашу думку, не пов’язаний із епідуральною анестезією та з мальформацією Кіарі і є проявом неконтрольованого перебігу епілепсії, оскільки хвора не отримувала протиепілептичних препаратів. Питання прийому протиепілептичних препаратів під час вагітності є дискутабельним, адже необхідно зважувати їх користь та потенційний шкідливий вплив на плід. Але враховуючи те, що судоми настали на третій день після операції, ми вважаємо, що породілля повинна обов’язково отримувати протиепілептичні препарати, так як відмова від прийому цих препаратів створює додатковий ризик для неї та для дитини – судоми можуть виникнути в будь-який момент, зокрема коли дитина буде на руках у матері, що може спричинити травмування як пацієнтки, так і її дитини.
Враховучи те, що аномалія Кіарі досить рідкісна та у літературі недостатньо даних з приводу анестезіологічного забезпечення кесарського розтину у цих пацієнток, необхідні подальші дослідження з приводу безпечності та переваг того чи іншого виду анестезії.
Повний перелік літератури знаходиться у редакції.
коментарів