

В первой части статьи (ЗТД №3/2013) были обсуждены вопросы дифференциальной диагностики анемий, сопровождающихся другими гематологическими аномалиями; а также анемий, сочетающихся с ретикулоцитозом
Если у ребенка с анемией нет других гематологических аномалий и отсутствует повышение уровня ретикулоцитов, следующий этап дифференциальной диагностики – оценка пациента с анемией и ретикулоцитопенией. При выявлении ретикулоцитопении, свидетельствующей о недостаточной продукции эритроцитов в костном мозге, направление дальнейшего диагностического поиска целесообразно определять, исходя из данных о размерах эритроцитов.
О размерах эритроцитов судят по показателю MCV (средний объем эритроцита, табл. 1). При оценке данного параметра следует учитывать, что объем эритроцитов изменяется с возрастом. У новорожденных показатель MCV находится в диапазоне 105–120 фл, у детей в возрасте от 6 мес. до 6 лет — 70–74 фл, 7–12 лет — 76–80 фл, у взрослых – около 80 фл. Некоторые исследователи предлагают рассчитывать нижнюю границу показателя MCV по формуле «70 + количество лет». То есть, у ребенка трех лет в норме средний объем эритроцита не должен быть меньше, чем 73 фл.
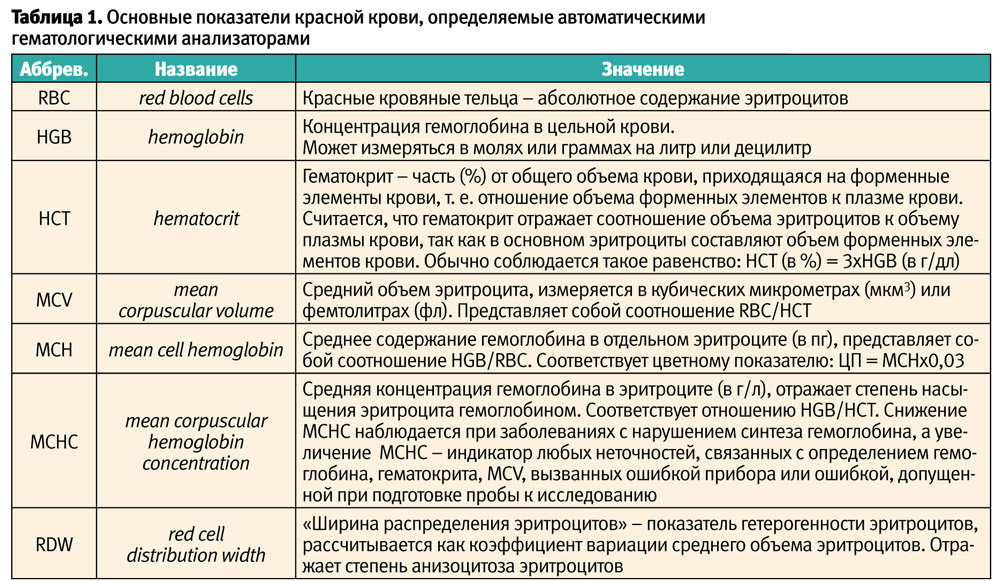
В зависимости от показателя MCV различают микроцитарные, нормоцитарные и макроцитарные анемии. О размерах эритроцитов можно судить и визуально, по микроскопической картине мазка периферической крови, но такая оценка более субъективна, а кроме того, сложнее соотносить размер эритроцитов с возрастом ребенка.
Анемия с ретикулоцитопенией и микроцитозом может быть вызвана дефицитом железа, дефектом синтеза эритроцитов, отравлением свинцом, а кроме того микроцитоз характерен для a-/b-талассемий и гемоглобин Е-синдромов (АЕ, ЕЕ).
Дефицит железа – самая частая причина анемий в детском возрасте. Пик заболеваемости приходится на детей от 6 месяцев до 3 лет, а также на подростков-девочек.
У детей первых месяцев жизни дефицит железа может развиться только вследствие кровопотери. Недостаточное поступление железа с пищей в этот период не имеет существенного значения, т. к. дети рождаются с определенным его запасом. Несмотря на то, что запас железа у детей, рожденных с низкой массой тела, недоношенных, детей из двоень, может быть меньше, чем у доношенных детей. Его обычно хватает до тех пор, пока масса ребенка не увеличится в два раза по сравнению с массой при рождении. Поэтому у доношенного ребенка младше полугода и у недоношенного младше четырех месяцев железодефицит, обусловленный исключительно недостаточным потреблением железа с пищей, маловероятен.
После истощения запасов основным источником железа становится пища. В этом возрасте основная причина дефицита железа – употребление в пищу продуктов с низким его содержанием – в первую очередь, коровьего молока. Употребление ребенком старше полугода более 700 мл коровьего молока в день – фактор возможного железодефицита. Наиболее часто железодефицитная анемия (ЖДА), обусловленная нерациональным вскармливанием, развивается в возрасте 9–24 мес. Именно с целью выявления нарушений в организации питания детей первого года жизни у нас, согласно приказу МЗУ № 149 от 20.03.08 г., всем здоровым детям в возрасте 9 мес. проводится плановое определение уровня гемоглобина в крови.
В каждом случае ЖДА в качестве возможной причины следует рассмотреть кровопотерю (см. начало статьи в № 3).
В клинической картине ЖДА, помимо симптомов анемии, присутствуют признаки сидеропенического синдрома (извращение вкуса, поперечная исчерченность ногтей, ухудшение памяти и обучаемости и др.).
В анализе периферической крови при ЖДА выявляют:
- снижение уровня гемоглобина и количества эритроцитов;
- микроцитоз (уменьшение MCV);
- гипохромию или снижение среднего содержания гемоглобина в эритроците (МСН) и средней концентрации гемоглобина в одном эритроците (МСНС);
- анизо- и пойкилоцитоз или увеличение RDW.
Биохимическим подтверждением наличия дефицита железа является:
- снижение уровня сывороточного ферритина;
- снижение уровня сывороточного железа;
- повышение железосвязывающей способности сыворотки (сывороточного трансферрина);
- снижение коэффициента насыщения трансферрина.
Сравнительно недавно была описана группа редких врожденных гипохромных микроцитарных анемий, рефрактерных к терапии препаратами железа (в англоязычной литературе IRIDA – iron refractory iron deficiency anemia). Одна из них связана с мутацией гена белка, участвующего в метаболизме железа – димерного транспортера металлов 1 (DMT-1). Она проявляется в раннем возрасте как ЖДА, не отвечающая на введение препаратов железа. При этом уровень ферритина, сывороточного железа и коэффициента насыщения трансферрина повышаются. Другая IRIDA, вызванная мутацией в гене TMPRSS6 (кодирует трансмембранную протеазу серина), была описана у многих пациентов. В основе ее лежит неадекватная гиперпродукция гепцидина, который ингибирует поступление железа в кровь из энтероцитов и выход железа из макрофагов.
Диагноз ЖДА требует обязательного лабораторного подтверждения дефицита железа! Ошибочный диагноз ЖДА и назначение препаратов железа пациентам с другими микроцитарными анемиями (сидеробластными, талассемиями) может привести к перегрузке железом и развитию тяжелых осложнений.
Сидеробластные (сидероахрестические) анемии обусловлены нарушениями синтеза гема врожденного и приобретенного генеза, при которых железо не встраивается в гем, а накапливается в митохондриях эритроцитов. Общими признаками всех сидеробластных анемий являются:
- наличие в периферической крови гипохромных микроцитарных эритроцитов, смешанных с нормальными эритроцитами, что создает картину диморфной популяции эритроцитов и приводит к увеличению показателя RDW;
- повышение концентрации сывороточного ферритина, сывороточного железа и насыщения железом трансферрина;
- появление в костном мозге кольцевых сидеробластов, которые представляют собой ядерные эритроциты с гранулами железа (агрегаты железа в митохондриях), расположенными вокруг ядра. Кольцевидные сидеробласты, являющиеся специфическим признаком сидеробластных анемий, следует отличать от нормальных сидеробластов, встречающихся в костном мозге здоровых людей. Нормальные сидеробласты являются предшественниками эритроцитов и содержат гранулы ферритина, равномерно рассеянные в цитоплазме.
В детском возрасте выявляются, в основном, врожденные сидеробластные анемии, возраст начала проявлений которых и степень выраженности симптомов значительно варьируют.
 Среди причин приобретенных сидеробластных анемий, в первую очередь, следует рассмотреть отравление свинцом. У детей со свинцовой интоксикацией могут иметь место анорексия, боли в животе, запор, рвота, а также поведенческие проблемы (импульсивность, невнимательность, гиперактивность), признаки периферической полинейропатии и повышения внутричерепного давления. Диагностика основывается на определении концентрации свинца в крови.
Среди причин приобретенных сидеробластных анемий, в первую очередь, следует рассмотреть отравление свинцом. У детей со свинцовой интоксикацией могут иметь место анорексия, боли в животе, запор, рвота, а также поведенческие проблемы (импульсивность, невнимательность, гиперактивность), признаки периферической полинейропатии и повышения внутричерепного давления. Диагностика основывается на определении концентрации свинца в крови.
Основным осложнением сидеробластных анемий является перегрузка железом. У пациентов с врожденной сидеробластной анемией даже в отсутствие значительного снижения уровня гемоглобина могут иметь место такие серьезные последствия перегрузки железом как сахарный диабет и цирроз печени.
Как уже указывалось выше (см. начало в №3), талассемии отмечаются в основном у представителей определенных этнических групп (жители Средиземноморья, Азии и Африки), поэтому в нашем регионе гомозиготные формы талассемий практически не встречаются. Тем не менее, учитывая рост числа детей, рожденных в смешанных этнических браках, вероятность появления гетерозиготных носителей генов талассемий увеличивается. Гетерозиготное носительство a- и b-талассемии проявляется умеренной микроцитарной анемией без развития типичного для гомозиготной талассемии гемолиза эритроцитов и эритробластоза.
Дифференциальная диагностика между ЖДА и гетерозиготным носительством талассемий и другими гемоглобинопатиями, особенно гемоглобинопатией Е, очень важна, т. к. в последних случаях назначение препаратов железа противопоказано. Простым отличительным признаком гемоглобинопатии Е является наличие нормального или несколько повышенного количества эритроцитов при сниженном уровне гемоглобина, тогда как при ЖДА снижается и уровень гемоглобина, и количество эритроцитов в крови. Анемия при гетерозиготном носительстве талассемий отличается от ЖДА отсутствием анизоцитоза и нормальными показателями RDW.
При носительстве b-талассемии микроцитарная анемия сочетается с повышенным уровнем гемоглобина А2 и/или фетального гемоглобина. Показатели обмена железа в норме, электрофорез не выявляет аномальных гемоглобинов.
Носительство a-талассемии характеризуется лишь наличием семейной умеренной микроцитарной анемии. Концентрация гемоглобина А2, фетального гемоглобина, показатели обмена железа и данные электрофореза гемоглобина – в норме. Носительство a-талассемии – диагноз исключения. Подтвердить его можно только при помощи молекулярно-генетических методов.
К анемиям с ретикулоцитопенией и нормоцитозом относят транзиторную эритробластопению детей, анемию хронического воспаления, анемию при хронической почечной недостаточности и анемию при гипотиреозе.
Транзиторная эритробластопения детей (ТЭД) – наиболее часто встречающаяся в детском возрасте приобретенная парциальная красноклеточная аплазия. Характеризуется медленно развивающейся анемией вследствие временного иммунного подавления красного ростка в костном мозге. ТЭД наблюдается у детей в возрасте от 6 мес. до 3 лет. Этиология заболевания неизвестна. Предполагается роль вирусной инфекции (но не парвовируса В19), т. к. заболеванию часто предшествует вирусная инфекция, хотя никаких определенных патогенов выявить не удалось. Не исключается также и генетическая предрасположенность. О том, что ТЭД имеет иммунологическую природу, свидетельствует тот факт, что сыворотка и гамма-глобулин пациентов с ТЭД способны подавлять рост эритроидных колоний in vitro. Восстановление продукции эритроцитов при ТЭД происходит спонтанно, без какого-либо лечения. Рецидивов не наблюдается. Все дети выздоравливают полностью и без осложнений.
Основное клиническое проявление ТЭД – постепенно, в течение нескольких недель, нарастающая бледность. Иногда родители могут отмечать слабость и утомляемость у ребенка.
При физикальном обследовании выявляются лишь признаки анемии (бледность кожи и слизистых, тахикардия, систолический шум при аускультации сердца) без какой-либо иной патологической симптоматики.
В общем анализе крови при ТЭД выявляется существенное снижение уровня гемоглобина — до 50–70 г/л, иногда – ниже, нормоцитоз, отсутствие ретикулоцитов или снижение их содержания. У 20–65% детей с ТЭД имеет место нейтропения. Нередко определяется умеренный тромбоцитоз. Причиной тромбоцитоза при красноклеточных аплазиях, также как и при ЖДА, является повышение продукции эритропоэтина (ЭПО), который структурно похож на тромбопоэтин (ТПО).
В пунктате костного мозга обнаруживается уменьшение количества ретикулоцитов и предшественников эритроцитов, хотя в типичных случаях ТЭД пункция костного мозга не является необходимой.
Длительность анемии при ТЭД обычно составляет 1–2 мес. О восстановлении продукции эритроцитов в костном мозге можно судить по появлению ретикулоцитоза в периферической крови.
ТЭД, в первую очередь, следует дифференцировать с анемией Блекфена–Даймонда, ЖДА и чистой красноклеточной аплазией, вызванной парвовирусной В19-инфекцией.
ТЭД от врожденной парциальной апластической анемии Блекфена–Даймонда отличает:
- более поздний возраст возникновения (после 6 мес.);
- отсутствие аномалий развития и задержки роста;
- соответствующий возрасту MCV;
- нормальный уровень фетального гемоглобина в крови;
- нормальный уровень активности аденозиндезаминазы эритроцитов.
В отличие от ЖДА, при ТЭД наблюдается:
- соответствующий возрасту MCV;
- нормальные показатели обмена железа; уровень сывороточного железа может быть повышенным из-за снижения интенсивности продукции эритроцитов.
Красноклеточная аплазия, вызванная парвовирусом В19 – возбудителем инфекционной эритемы – в большей или меньшей степени имеет место у всех заболевших. Это связано с тропностью и цитотоксичностью парвовируса В19 в отношении предшественников эритроцитов в костном мозге. Но поскольку течение парвовирусной инфекции непродолжительное, у детей, в остальном здоровых, с нормальной длительностью жизни эритроцитов, клинически значимая анемия не успевает развиться или проходит незамеченной. Но у детей с хроническим гемолизом, у которых длительность жизни эритроцитов укорочена, временное подавление красного ростка костного мозга во время парвовирусной инфекции может приводить к развитию тяжелой анемии – т. н. апластическому кризу.
Снижение уровня гемоглобина при апластическом кризе происходит быстро – в течение нескольких дней. При этом присутствуют признаки инфекционной эритемы («отшлепанные» щеки, кружевная сыпь на теле) и симптомы предшествовавшего хронического гемолиза (увеличение селезенки, субиктеричность склер, признаки эритроидной гиперплазии). Падение уровня гемоглобина во время апластического криза не сопровождается нарастанием непрямой билирубинемии.
Апластический криз, вызванный парвовирусом В19, у пациентов с хроническим гемолизом может развиться только один раз в жизни. Однако работа костного мозга может нарушаться другими вирусными инфекциями.
В редких случаях, у пациентов с тяжелым иммунодефицитом (ВИЧ-инфекция, врожденные ИДЗ, медикаментозная иммуносупрессия) может наблюдаться персистенция парвовируса В19, которая, в свою очередь, приводит к развитию чистой красноклеточной аплазии с выраженной анемией. Такая анемия может приниматься за ТЭД. Но в отличие от ТЭД, при чистой красноклеточной аплазии спонтанного восстановления активности костного мозга не происходит, пациентам требуются неоднократные переливания эритроцитарной массы. Диагноз в этом случае основывается на обнаружении частиц вируса методом ПЦР (серологические тесты непригодны, т. к. синтез антител снижен).
Вообще, при ТЭД может возникать необходимость в переливании эритроцитарной массы, но если пациентам с возможной ТЭД потребовалось более одного переливания, следует искать другие причины анемии.
У детей первых месяцев жизни дефицит железа может развиться только вследствие кровопотери
Причиной развития анемии при хронических заболеваниях, как предполагают, является постоянная продукция провоспалительных цитокинов (ИЛ-1 и ФНО-а), которая через каскад реакций приводит к перераспределению пула железа, недостаточной продукции ЭПО и снижению ответа костного мозга на ЭПО. К таким хроническим заболеваниям относят хронические гноеродные инфекции (хронический остеомиелит, бронхоэктазы), хронические воспалительные заболевания кишечника, диффузные болезни соединительной ткани.
Чаще всего анемию хронических заболеваний приходится дифференцировать с ЖДА, тем более что железодефицит при хронических болезнях тоже может иметь место.
Основными отличиями анемии хронических заболеваний от ЖДА являются:
- нормоцитоз (хотя иногда может определяться и небольшое количество микроцитов);
- несмотря на снижение уровня сывороточного железа, уровень ферритина в норме или повышен, железосвязывающая способность снижена;
- концентрация растворимых рецепторов трансферрина в норме, тогда как при ЖДА она повышена. Это наиболее важный дифференциально-диагностический критерий.
Анемия при хронической почечной недостаточности, в целом, имеет те же механизмы развития, что и анемия хронического воспаления, но добавляется прогрессирующее снижение продукции ЭПО. Принципы дифференциальной диагностики не отличаются от таковых при анемии хронического воспаления.
При выявлении анемии с ретикулоцитопенией и макроцитозом, следует выяснить, имеются ли черты мегалобластного кроветворения – гиперсегментированные нейтрофилы, тельца Кебота и Жолли в эритроцитах. Мегалобластные анемии почти всегда развиваются вследствие дефицита витамина В12 и/или фолиевой кислоты. К макроцитарным анемиям без признаков мегалобластного кроветворения относятся анемия Блекфена–Даймонда, врожденные дизэритропоэтические анемии и синдром Пирсона.
Основными критериями диагностики врожденной парциальной апластической анемии (анемии Блекфена–Даймонда) являются: выявление анемии в раннем младенческом возрасте (в 90% случаев – на первом году жизни, в среднем – в возрасте 3 мес.), макроцитоз, ретикулоцитопения, а также дефицит или отсутствие предшественников эритроцитов в костном мозге при нормальной клеточности других ростков. В основе заболевания лежит генетический дефект предшественников эритроцитов, обуславливающий их повышенный апоптоз.
У приблизительно 50% детей с анемией Блекфена–Даймонда имеются врожденные аномалии развития (черепно-лицевой дизморфизм, аномалии конечностей, например, трехфаланговые большие пальцы) и низкорослость.
При лабораторных исследованиях выявляется значительное повышение MCV, нередко выше 98 фл. Вообще, уровень MCV выше 98 фл вне периода новорожденности всегда указывает на серьезную гематологическую проблему вроде анемии Блекфена–Даймонда, миелодиспластического синдрома и лейкемии, или на врожденное нарушение метаболизма. Признаки мегалобластного кроветворения при анемии Блекфена–Даймонда отсутствуют. Вследствие повышенной продукции ЭПО может отмечаться тромбоцитоз. Уровень фолиевой кислоты и витамина В12 в сыворотке нормальный, железа – повышенный. Повышены уровень фетального гемоглобина и активность аденозиндезаминазы эритроцитов. Анализ хромосом на разрывы отрицательный.
Костномозговой и панкреатический синдром Пирсона характеризуется макроцитарной анемией, развивающейся уже в неонатальном периоде, иногда – с нейтропенией и тромбоцитопенией, повышением уровня фетального гемоглобина, наличием в костном мозге вакуолизированных эритробластов и миелобластов, а также задержкой развития, панкреофиброзом с развитием эндо- и экзокринной недостаточности поджелудочной железы, мышечными и неврологическими расстройствами. Анемия при синдроме Пирсона – сидеробластная, о чем свидетельствует обнаружение кольцевых сидеробластов в костном мозге, но в отличие от других сидеробластных анемий – макроцитарная.
Врожденные дизэритропоэтические анемии – это группа заболеваний, при которых в костном мозге производятся эритроциты с аномальной структурой, что клинически проявляется анемией разной степени при повышенной эритроидной активности костного мозга (неэффективный эритропоэз). Как правило, присутствует умеренный гемолиз (повышение уровня непрямого билирубина, снижение уровня гаптоглобина в сыворотке), но без ретикулоцитоза, а также спленомегалия.
Фолиеводефицитная анемия чаще развивается на первом году жизни. Запасы фолиевой кислоты в организме скудные, поэтому клинические проявления фолиеводефицитной анемии развиваются уже через 2–3 месяца после прекращения поступления фолатов с пищей.
Данная анемия мегалобластная, нередко сочетается с нейтропенией (большие нейтрофилы с гиперсегментированными ядрами) и тромбоцитопенией. В крови определяются снижение уровня фолиевой кислоты и повышение уровня ЛДГ. Более надежным показателем дефицита фолатов является снижение уровня фолатов в эритроцитах. В костном мозге – гиперклеточность из-за эритроидной гиперплазии, предшественники эритроцитов имеют мегалобластные изменения, хотя встречаются и нормальные клетки.
Причины развития дефицита фолатов у детей:
- недостаточное поступление фолатов с пищей – вскармливание козьим, порошковым молоком, дефицит фолатов у матери во время беременности;
- вторичный дефицит фолатов у пациентов с хроническим гемолизом;
- нарушение всасывания фолатов – целиакия, хронический инфекционный энтерит, межкишечная фистула, резекция кишечника, прием антиконвульсантов;
- врожденная аномалия обмена фолатов – дефицит дигидрофолатредуктазы;
- нарушения обмена фолатов, вызванные приемом лекарственных средств – метотрексата, пириметамина, триметоприма.
Анемия, связанная с дефицитом витамина В12, нередко сочетается с такими признаками как глоссит, рвота, диарея, желтуха. Нередко отмечается неврологическая симптоматика – парестезии, нарушения чувствительности, мышечная гипотония, судороги, задержка ПМР, нейропсихические нарушения. Неврологические проявления дефицита витамина В12 могут отмечаться и без гематологических аномалий.
Лабораторным подтверждением дефицита витамина В12 является выявление мегалобластной анемии (в тяжелых случаях – с нейтропенией и тромбоцитопенией), снижение уровня витамина В12 в сыворотке крови, повышение активности сывороточной ЛДГ, а также повышенная экскреция с мочой метилмалоновой кислоты.
Причинами развития дефицита витамина В12 могут быть:
- недостаточное поступление с пищей (очень редко) – у строгих вегетарианцев;
- отсутствие внутреннего фактора:
- врожденная пернициозная анемия – редкое аутосомно-рецессивное заболевание, при котором отсутствует продукция внутреннего фактора (ВФ) в желудке или синтезируется аномальный ВФ. Отличается от заболевания взрослых отсутствием антител к париетальным клеткам желудка и соответствующих эндокринных нарушений. Симптомы проявляются к возрасту около 1 года;
- ювенильная пернициозная анемия – проявление аутоиммунного полигландулярного синдрома (хронический слизисто-кожный кандидоз, гипопаратиреоз, аутоиммунный атрофический гастрит и др.);
- последствие операций на желудке;
- нарушение всасывания витамина В12 – неонатальный некротизирующий энтероколит, болезнь Крона, удаление терминальной части подвздошной кишки, чрезмерный рост кишечных бактерий в дивертикулах и дупликатурах тонкого кишечника, глистная инвазия (дифиллоботриоз), генетический дефект рецептора, связывающего комплекс «витамин В12+ВФ» (синдром Имерслунд–Гренсбека);
- отсутствие транспортного белка витамина В12 – дефицит транскобаламина ІІ (проявляется тяжелой мегалобластной анемией и другими клиническими проявлениями дефицита витамина В12 с первых недель жизни, но при этом уровень витамина В12 в плазме нормальный).
Таким образом, использование представленного пошагового алгоритма диагностики и дифференциальной диагностики, на наш взгляд, позволит диагностировать или заподозрить большинство из возможных причин анемий у детей уже на амбулаторном этапе обследования. А применение автоматических гематологических анализаторов с расчетом эритроцитарных индексов будет способствовать повышению эффективности диагностики анемий.
Литература:
- Берман Р. И., Клигман Р. М., Дженсон Х. Б. Педиатрия по Нельсону. Том 4. – М.: Рид Элсивер, 2009. – С. 824-906.
- Луговская С. А. Что могут гематологические анализаторы // Лаборатория. – 1997. – № 5. – С. 22-25.
- Inoue S. Pediatric Chronic Anemia. – Режим доступа: http://emedicine.medscape.com/article/954598-overview.
- Huang L. H. Transient Erythroblastopenia of Childhood. – Режим доступа: http://emedicine.medscape.com/article/959644-overview.
- Badawy M. K. Pediatric Lead Toxicity. Режим доступа: http://emedicine.medscape.com/article/1009587-overview.
коментариев