

(Окончание. Начало – в №1 (46) 2014)
Криопирин-ассоциированные периодические синдромы (Cryopyrin-Associated Periodic Syndromes – CAPS) связаны с мутациями в гене NLRP3 (CIAS1), кодирующем синтез белка криопирина (NALP3/PYPAF1). Ген NLRP3 (CIAS1) локализован на длинном плече 1-й пары хромосом (1q44) и экспрессируется в полиморфноядерных лейкоцитах, моноцитах и хондроцитах. Криопирин – основной компонент NLRP3-инфламмасомы – цитоплазматического белкового комплекса, запускающего воспалительную реакцию при контакте фагоцитов с микроорганизмами (рис. 1).
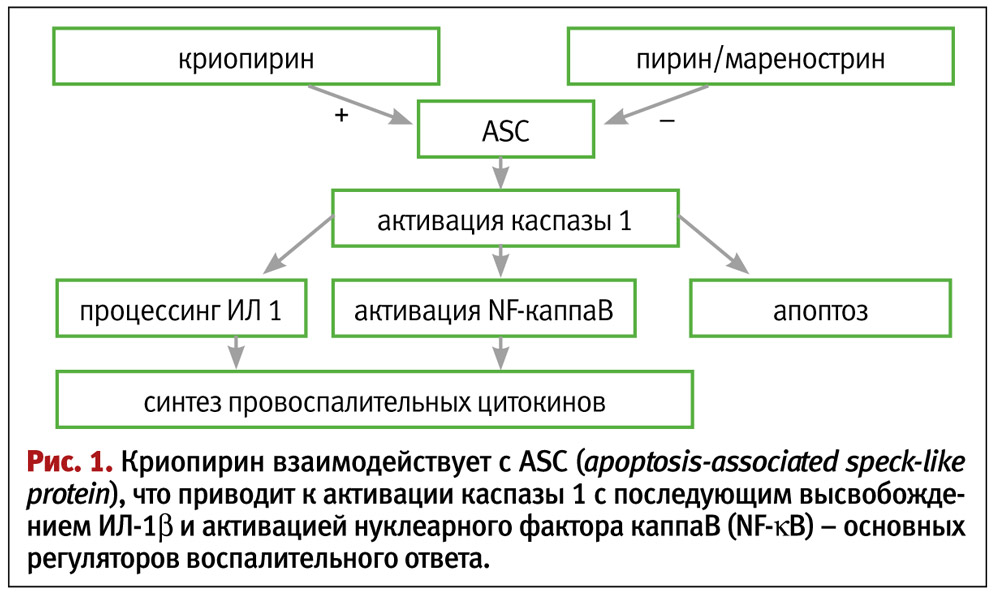
Инфламмасомы – важный компонент системы врожденного иммунитета, которая основана на клеточной системе быстрого распознавания генетически инородных структур и их уничтожения. Распознавание патогенов осуществляется мембранными Тoll-подобными рецепторами и цитозольными NOD-подобными рецепторами. Некоторые NOD-подобные рецепторы распознают не только инфекционные сигналы, но и т. н. немикробные «сигналы опасности». В частности, известными лигандами криопирина являются бактериальная и вирусная РНК, а также кристаллы мочевой кислоты. При связывании лиганда криопирин взаимодействует с протеином ASC, что приводит к активации каспазы 1, и в дальнейшем – к внутриклеточному процессингу и образованию зрелой активной формы интерлейкина 1b.
При криопирин-ассоциированных периодических синдромах (CAPS) мутации в гене NLRP3 (CIAS1) приводят к постоянной избыточной продукции ИЛ-1b, тогда как в норме его синтез увеличивается только в ответ на инфекцию.
 Известны 3 формы CAPS:
Известны 3 формы CAPS:
1) семейный холодовой аутовоспалительный синдром (Familial Cold Autoinflammatory Syndrome – FCAS) – впервые описан в 1940 г.;
2) синдром Макл–Уэлса (Muckle–Wells Syndrome – MWS) – впервые описан в 1962 г.;
3) хронический младенческий нервно-кожно-артикулярный синдром/ младенческое мультисистемное воспалительное заболевание (Chronic Infantile Onset Neurologic Cutneous Articular/Neonatal Onset Multisystem Inflammatory Disease – CINCA/NOMID) – впервые описан в 1981 г.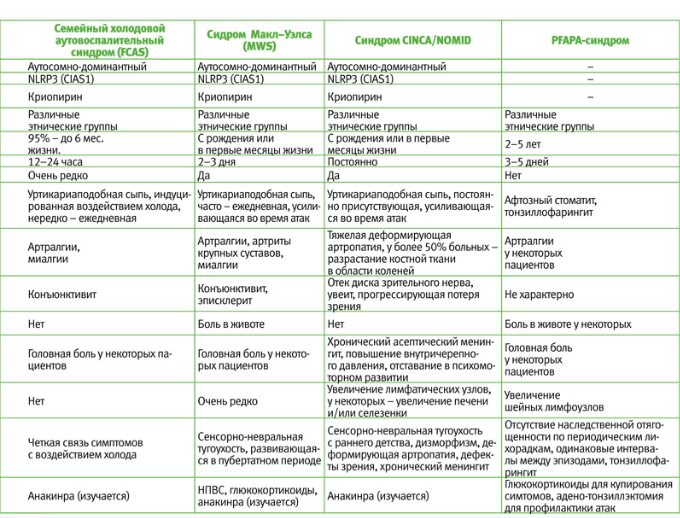
Для всех этих заболеваний характерны:
- раннее начало (на первом году жизни, иногда – позже);
- наличие рецидивирующей или постоянной лихорадки, уртикариаподобной сыпи, поражения суставов различной степени, конъюнктивита и головной боли;
- аутосомно-доминантный тип наследования.
В настоящее время идентифицированы 111 мутаций в гене NLRP3 (CIAS1), приводящих к развитию криопиринопатий. FCAS, MWS и CINCA/NOMID были описаны в разное время и ранее считались самостоятельными нозологическими единицами. Однако, после того, как были выявлены общие патогенетические механизмы и генетическая основа этих заболеваний, их объединили в одну группу – CAPS, и в настоящее время считают различными клиническими вариантами одного заболевания. При этом FCAS – легкая форма с благоприятным прогнозом, MWS протекает тяжелее с возможностью развития необратимых изменений в некоторых органах (тугоухость, амилоидоз), а CINCA/NOMID – наиболее тяжелый вариант криопиринопатий, при котором упорное воспаление быстро приводит к необратимым изменениям в большинстве органов и систем организма.
Сыпь – наиболее ранний и постоянный признак CAPS. У большинства пациентов сыпь появляется уже при рождении или вскоре после него. В некоторых случаях MWS и FCAS сыпь может появиться в более позднем возрасте. Макулопапулярная, напоминающая крапивницу, сыпь покрывает все тело и усиливается в периоды воспалительных атак. Чаще всего сыпь не зудит, но некоторые пациенты жалуются на зуд или даже жжение. При биопсии кожи в области элементов сыпи выявляют увеличение количества нейтрофилов вокруг спиралевидных выводных протоков эккринных потовых желез и кровеносных сосудов. У некоторых пациентов, как правило, с FCAS, сыпь отмечается только во время воспалительных атак, но у большинства больных сыпь имеет место практически каждый день, во время атак значительно усиливаясь.
Семейный холодовой аутовоспалительный синдром (FCAS), известный также как семейная холодовая крапивница (familial cold urticaria – FCU) и холодовая гиперчувствительность, характеризуется появлением сыпи, артралгий, конъюнктивита и лихорадки после воздействия холода. FCAS отличается от других наследственных синдромов периодической лихорадки наличием четкого провоцирующего фактора – холода, ранним началом (обычно до шестимесячного возраста) и небольшой продолжительностью эпизодов – менее 24 часов.
У 95% пациентов приступы начинаются в течение первых 6 месяцев, у 60% – в течение первых нескольких дней жизни. Средний возраст начала симптомов – 47 дней, с диапазоном от 2 часов до 10 лет. Тяжесть течения не зависит от возраста начала симптомов. Сыпь максимально выражена в подростковом возрасте и в юности.
Воспалительные атаки при FCAS всегда связаны с воздействием холода. При этом диапазон температур, способных вызвать симптомы заболевания, и длительность воздействия значительно варьируют. У одних пациентов симптомы появляются после серьезного переохлаждения (например, в холодные ветреные дни), а у других – после совсем незначительных воздействий (употребление холодной пищи или напитков, плавание, пребывание вблизи кондиционера).
Симптомы появляются через 1–2 часа после воздействия холода. Помимо сыпи, обычно отмечается лихорадка с ознобом (93%), полиартралгии (96%) и конъюнктивит (84%). Реже в период приступа у пациентов наблюдается обильное потоотделение (78%), сонливость (67%), головная боль (58%), неукротимая жажда (53%), тошнота (51%) и миалгии. Развитие артритов не характерно для воспалительных атак при FCAS. Средняя длительность приступа – 12 часов (от 30 минут до 72 часов), у 94% пациентов большинство приступов заканчивается менее чем за 24 часа.
Диагностические критерии семейного холодового аутовоспалительного синдрома – FCAS (Hoffman HM et al., 2001)
- Рецидивирующие эпизоды лихорадки и сыпи, возникающие после воздействия холода (естественного и/или экспериментального).
- Аутосомно-доминантный тип наследования.
- Возраст начала заболевания – менее 6 месяцев жизни.
- Продолжительность большинства приступов менее 24 часов.
- Конъюнктивит, связанный с приступами.
- Отсутствие глухоты, периорбитального отёка, увеличения лимфоузлов и серозитов.
FCAS следует отличать, в первую очередь, от приобретенной холодовой крапивницы – наиболее частой формы физической крапивницы, при которой симптомы обусловлены дегрануляцией тучных клеток с высвобождением гистамина. В отличие от FCAS, приобретенная холодовая крапивница обычно возникает у взрослых и спонтанно разрешается. Сыпь при приобретенной холодовой крапивнице возникает непосредственно после прямого воздействия холода (в течение нескольких минут), проходит в течение нескольких часов и может сопровождаться ангионевротическим отеком, бронхиальной обструкцией и гипотензией.
Кроме того, проводится дифференциальная диагностика с другими синдромами периодической лихорадки, криоглобулинемией и крапивницей.
Синдром Макл–Уэлса (MWS) – более тяжелый вариант криопиринопатии, при котором помимо сыпи, лихорадки и конъюнктивита, в период воспалительных атак отмечается артрит и боль в животе. У части пациентов с возрастом развивается сенсорная тугоухость.
Воспалительные атаки при синдроме Макл–Уэлса длятся 24–72 часа и чаще всего провоцируются холодом, сыростью, стрессом, физическими нагрузками и неизвестными факторами. При этом связь приступов с воздействием холода не так очевидна, как при FCAS. Суставной синдром при MWS может варьировать от непродолжительных артралгий до полиартритов с вовлечением, как правило, крупных суставов. Нейросенсорная тугоухость развивается у 50–70% пациентов, обычно в подростковом возрасте.
После нескольких лет заболевания у 20–40% больных MWS развивается системный амилоидоз, тогда как при FCAS описаны лишь единичные случаи этого осложнения.
Дифференцировать синдром Макл–Уэлса нужно не только с другими аутовоспалительными заболеваниями, но и нередко с синдромом Альпорта, при котором, как известно, имеют место тугоухость, поражение глаз и почечная недостаточность.
Синдром CINCA/NOMID – наиболее тяжелая форма CAPS, характеризующаяся персистирующим выраженным воспалением с поражением кожи, костей, суставов и ЦНС.
В отличие от других синдромов периодической лихорадки, при синдроме CINCA/NOMID лихорадка присутствует практически ежедневно, нередко с первых дней жизни, или же могут отмечаться непродолжительные, но очень частые эпизоды лихорадки. Также постоянным симптомом является уртикариаподобная сыпь, которая может менять локализацию, быть более или менее выраженной, но присутствует практически все время от начала заболевания.
Поражение ЦНС у пациентов с синдромом CINCA/NOMID характеризуется развитием хронического асептического менингита, вызванного нейтрофильной инфильтрацией мозговых оболочек, который проявляется, в первую очередь, головными болями и раздражительностью. В дальнейшем присоединяются симптомы повышения внутричерепного давления (отек дисков зрительных нервов, наружная и внутренняя гидроцефалия, атрофия мозга). У большинства детей с синдромом CINCA/NOMID уже в раннем детстве формируется нейросенсорная тугоухость. Нередко дети отстают в умственном развитии и страдают от симптоматической эпилепсии.
Стойкие изменения суставов появляются в течение первых лет жизни в виде полиартрита с симметричным поражением крупных суставов. Характерен усиленный рост метафизов и эпифизов длинных трубчатых костей, надколенника, а также ускорение оссификации ядер окостенения, что приводит к серьезным деформациям и контрактурам в конечностях, нарушению их функции и болевому синдрому.
Поражение глаз при синдроме CINCA/NOMID характеризуется конъюнктивитом, передним (50%) или задним (20%) увеитом, атрофией зрительного нерва (25%). Больные имеют дизморфические особенности: седловидную переносицу, макроцефалию, нависающий лоб, низкий рост с короткими утолщенными конечностями, контрактуры суставов (особенно, коленных). Нередко определяется увеличение лимфатических узлов и селезенки.
Синдром CINCA/NOMID дифференцируют с другими аутовоспалительными синдромами, а также с болезнью Стилла. Отличием поражения суставов при синдроме CINCA/NOMID является формирование суставных контрактур за счет разрастания костной ткани, а не утолщения синовиальных оболочек и периартикулярных тканей, что имеет место при болезни Стилла.
Диагностика криопиринопатий
Диагностика криопиринопатий базируется, в первую очередь, на клинико-анамнестических данных. Наличие характерного симптомокомплекса в сочетании с указанием на аналогичные симптомы у ближайших родственников пациентов в нескольких поколениях имеет место у большинства больных с FCAS и MWS. При синдроме CINCA/NOMID более чем в половине случаев наследственность не отягощена, заболевание вызвано мутацией de novo.
В период воспалительных атак в крови повышаются уровни острофазовых показателей (лейкоциты, СОЭ, СРБ), при синдроме CINCA/NOMID может также отмечаться анемия, повышение уровня эозинофилов в крови и спинномозговой жидкости, гиперглобулинемия. Для верификации патогномоничных для синдрома CINCA/NOMID изменений костей и суставов проводится рентгенография коленей. С целью подтверждения диагноза FCAS может быть проведена холодовая проба: к коже на 5 минут прикладывают кубик льда, после чего фиксируют характерные для FCAS изменения. Кроме того, в западных странах доступно генетическое исследование, подтверждающее наличие мутаций в гене NLRP3 (CIAS1).
До недавнего времени в терапии криопиринопатий с небольшим успехом применяли глюкокортикоиды, колхицин, цитостатики. При этом пациенты с синдромом CINCA/NOMID погибали еще в детском возрасте. Однако в настоящее время возможности лечения криопиринопатий значительно расширились – применение рекомбинантных антагонистов рецепторов ИЛ-1 (рилонацепт, канакинумаб, анакинра) позволяет эффективно контролировать симптомы заболеваний.
Синдром PFAPA (Periodic Fever, Aphthous Stomatitis, Pharyngitis, Adenitis Syndrome) впервые был описан G. S. Marshall в 1987 на основании наблюдения 12 детей с периодической лихорадкой, сопровождавшейся фарингитом, афтозным стоматитом и увеличением шейных лимфоузлов. Первоначально этот синдром был назван «синдромом Маршалла», но позднее более распространенным названием стала аббревиатура, отражающая основные клинические проявления синдрома – «PFAPA».
В отличие от других синдромов периодической лихорадки, синдром PFAPA является спорадическим заболеванием, никаких данных, позволяющих предположить его наследственный характер, пока не получено. Этиология неизвестна. Есть данные, что в период приступов лихорадки в крови отмечается повышение уровней провоспалительных цитокинов: ИЛ1b, ТNFa, ИЛ6 и ИЛ12р70. При этом в межприступный период полной нормализации этих показателей не наблюдается, что указывает на то, что воспалительный процесс персистирует.
Болеют чаще всего дети в возрасте от 2 до 5 лет, с небольшим преобладанием мальчиков. Никаких этнических особенностей нет. Синдром PFAPA – самый распространенный из синдромов периодической лихорадки, он встречается гораздо чаще, чем моногенные аутовоспалительные синдромы.
Эпизоды лихорадки при PFAPA-синдроме длятся от 3 до 5 дней и повторяются каждые 3–5 недель. Температура тела повышается резко, нередко до 40–41°С, лихорадка сопровождается ознобом. Небольшие (менее 1 см в диаметре) афты на слизистой оболочке полости рта или на губах появляются в течение 12–24 часов от начала лихорадки, они умеренно болезненны и в течение недели заживают полностью без образования рубцов. В большинстве случаев также отмечается двустороннее увеличение шейных лимфоузлов и тонзиллофарингит, обычно без формирования налетов на миндалинах. Другие, менее распространенные симптомы – боль в животе, артралгии и головная боль.
В период лихорадки у большинства детей в крови определяется повышение СОЭ, количества лейкоцитов и уровня С-реактивного белка. В посевах мазков с миндалин, взятых во время эпизодов лихорадки, b-гемолитический стрептококк группы А отсутствует. Увеличение концентрации IgD иногда может иметь место, но оно не столь значительно, как при HIDS.
В межприступном периоде дети с синдромом PFAPA хорошо себя чувствуют, никаких отклонений при клиническом и лабораторном обследовании не выявляется, никакого негативного воздействия на рост и развитие детей воспалительные атаки не имеют.
Прогноз при синдроме PFAPA весьма благоприятный. У большинства детей после 5–7-летнего возраста симптомы полностью проходят. Лишь у небольшого числа пациентов старше 7 лет после того, как классический клинический симптомокомплекс перестал появляться, еще в течение нескольких лет могут отмечаться редкие и нерегулярные неклассические эпизоды.
Критерии диагностики синдрома PFAPA (K. T. Thomas et al., 1999)
Наличие всех 3 критериев из следующих:
1) 3 и более документированных эпизода лихорадки, длившихся не более 5 дней, с равномерными интервалами между эпизодами длительностью от 3 до 6 недель;
2) наличие во время эпизодов лихорадки шейной лимфаденопатии, тонзиллофарингита и/или афтозного стоматита;
3) нормальное состояние между эпизодами, отсутствие нарушений роста и развития.
Критерии, исключающие PFAPA:
1) нейтропения во время эпизода лихорадки или непосредственно предшествующая ему;
2) атипичная симптоматика;
3) повышение острофазовых показателей между эпизодами;
4) указания на периодические лихорадки в семейном анамнезе.
Единого мнения относительно лечения детей с синдромом PFAPA нет. Учитывая доброкачественный характер его течения, спонтанное разрешение с возрастом, приходится выбирать лишь те лечебные мероприятия, при которых риск побочных эффектов минимален. Лечебная тактика при синдроме PFAPA преследует 2 цели: 1) купирование симптомов во время эпизодов лихорадки; 2) предупреждение новых атак заболевания.
Нестероидные противовоспалительные препараты и парацетамол при PFAPA-синдроме недостаточно эффективны. Назначение глюкокортикоидов (от 0,5 мг/кг до 1–2 мг/кг в сутки по преднизолону) коротким курсом в начале атаки способствует быстрому купированию симптомов. Однако, применение кортикостероидов при каждом лихорадочном эпизоде способствует сокращению интервала между атаками.
Как показывают проведенные недавно рандомизированные контролируемые исследования, наиболее эффективной мерой профилактики воспалительных атак при синдроме PFAPA является аденотонзиллэктомия.
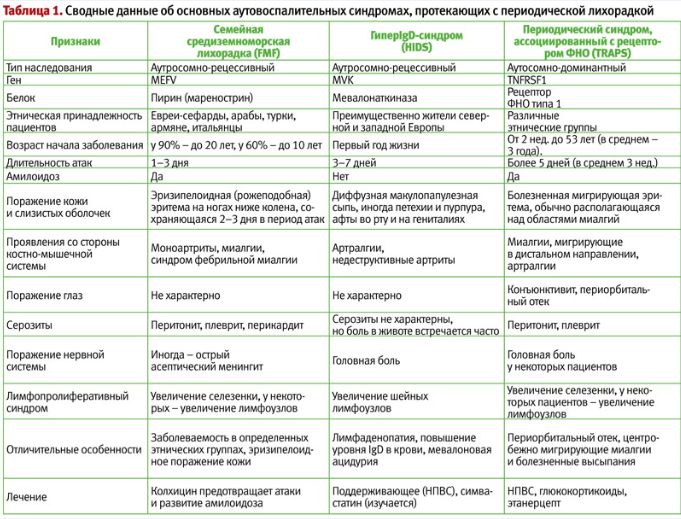
Дифференциальная диагностика синдромов периодической лихорадки основывается, в первую очередь, на клинико-анамнестических данных (табл. 1), т. к. генетическое исследование дорого и доступно лишь в немногих центрах. При этом принципиально важно отличать синдром PFAPA от моногенных синдромов периодической лихорадки (особенно, FMF, HIDS и TRAPS), поскольку последние имеют не такой благоприятный прогноз, как синдром PFAPA, и требуют иных подходов к лечению. С другой стороны, критерии диагностики синдрома PFAPA низкоспецифичны. Как показывают исследования M. Gattorno, под диагностические критерии синдрома PFAPA попадают 83% больных с гипер IgD-синдромом, 57% больных TRAPS и 8% больных семейной средиземноморской лихорадкой. В связи с этим, была предложена т. н. шкала Gaslini, включающая признаки, наличие которых указывает на вероятность моногенного синдрома периодической лихорадки, что, в свою очередь, требует проведения генетических тестов. Эти признаки включают:
- начало заболевания в раннем возрасте;
- наличие периодической лихорадки в семейном анамнезе;
- боль в животе/груди;
- диарею;
- афтозный стоматит.

Заключение
Аутовоспалительные синдромы – недавно сформировавшееся направление в современной ревматологии, но интерес к ним неуклонно растет. По мере ознакомления медицинской общественности с клинико-генетическими особенностями аутовоспалительных синдромов, и в том числе тех, что проявляются периодической лихорадкой, эта патология диагностируется все чаще.
Первые симптомы большинства этих заболеваний появляются в детском возрасте, поэтому именно для педиатров важно уметь заподозрить аутовоспалительный синдром. Еще большее значение своевременной диагностике аутовоспалительных синдромов придает тот факт, что в настоящее время, благодаря использованию генно-инженерных препаратов, способных ингибировать медиаторы системы врожденного иммунитета, в лечении данной патологии наметился прорыв, когда удается полностью контролировать симптомы заболеваний, считавшихся ранее инвалидизирующими или даже смертельными.
Наличие у ребенка 2 или 3 документированных эпизодов лихорадки в сочетании с характерными (описанными выше) клиническими и лабораторными данными должно стать поводом для направления к детскому ревматологу и/или инфекционисту для уточнения диагноза и дальнейшего обследования. Диагностика аутовоспалительных синдромов, протекающих с периодической лихорадкой, требует исключения инфекционной, аутоиммунной, онкологической патологии и врожденных иммунодефицитных заболеваний. Значительным подспорьем в диагностике синдромов периодической лихорадки стали генетические исследования, которые становятся все более доступными для многих стран благодаря международным программам (проект EUROFEVER).
Список литературы находится в редакции.
коментариев