

Аутоиммунный лимфопролиферативный синдром (ALPS – autoimmune lymphoproliferative syndrome) – редкое заболевание, относящееся к первичным иммунодефицитам, в основе которого лежит генетический деффект апоптоза (запрограмированной смерти лимфоцитов). В клиническом аспекте данное заболевание характеризуется лимфопролиферативным синдромом и аутоиммунными цитопениями с высоким риском развития лимфомы. В мире описано около 500 случаев ALPS
Этиология и патогенез
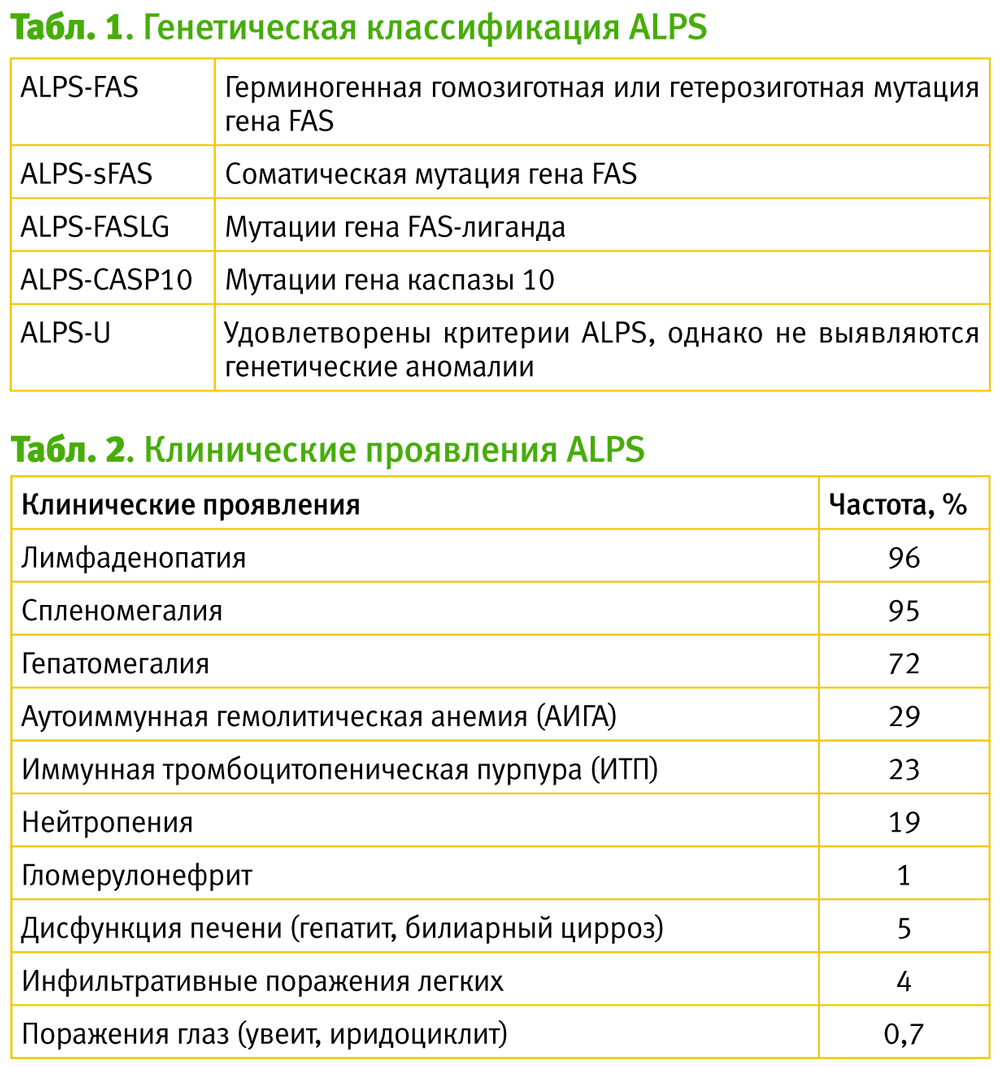
Основным этиологическим фактором служит герминогенная или соматическая мутация в генах, кодирующих FAS-рецептор, FAS-лиганд (FASLG) каспазы-10. Примерно у 1\3 пациентов с ALPS генетический деффект остается неизвестным (см табл. 1). Указанный дефект FAS-опосредованного апоптоза приводит к потере контроля над пролиферацией лимфоцитов, вследствие чего происходит накопление активированных лимфоцитов в лимфоидных органах, что выражается развитием лимфаденопатии и спленомегалии, а также формированием аутоиммунного ответа.
Клинические проявления
Клиническая картина ALPS разнообразна и включает в себя синдромы лимфоидной пролиферации и аутоиммунных нарушений (см. табл. 2).
Наиболее ранним и самым распространенным клиническим проявлением ALPS является хроническая лимфаденопатия и\или спленомегалия, которые часто являются бессимптомными и выявляются случайно во время рутинного обследования. Пациенты с ALPS также могут иметь мультилинейную цитопению, которая является хронической и может быть рефрактерной к терапии.
ALPS может манифестироваться эпизодами выраженной слабости, бледности, иктеричности вследствие гемолитической анемии, геморрагическим синдромом из-за тромбоцитопении, а также бактериальными инфекциями как следствие нейтропении.
Критерии диагностики ALPS
Перечень диагностических критериев включает в себя 2 обязательных и 6 дополнительных клинических и лабораторных признаков. Среди дополнительных выделяют первичные и вторичные (см. табл. 3).
Окончательный (достоверный) диагноз ALPS должен включать оба основных критерия и один дополнительный первичный; вероятный диагноз – оба основных критерия и один дополнительный вторичный критерий.
 Дифференциальная диагностика
Дифференциальная диагностика
Необходимо, прежде всего, провести дифференциальную диагностику с аутоиммунными, ревматологическими, инфекционными и онкологическими заболеваниями.
Всем пациентам в начале диагностики должна быть проведена биопсия лимфатического узла для исключения в первую очередь лимфомы (неходжкинской или ходжкинской).
Далее диагностический поиск предлагает исключить более редкие заболевания, такие как:
- - Болезнь Каслмана (Castleman disease) – ангиофолликулярная гиперплазия лимфоузлов;
- - Болезнь Дункана – Х-сцепленное Т- и NK-клеточное лимфопролиферативное заболевание;
- - DALD-синдром (Dianzani autoimmune lymphoproliferative disease)– связан с аномалией гена в хромосомном участке 4q21-q25);
- - Гипер-IgM-синдром;
- - Синдром Вискотта–Олдрича;
- - Синдром Эванса;
- - Болезнь Розаи–Дорфмана – синусовый гистиоцитоз с массивной лимфаденопатией;
- - Болезнь Кучи–Фудзимото — гистиоцитарный некротизирующий лимфаденит.
Также необходимо помнить о ALPS-подобных заболеваниях: с аномалиями генов каспазы-8, NRAS, KRAS и других.
Лечение
Основной части пациентов, получившей диагноз на этапе лимфоидной пролиферации, специфическое лечение не показано. Терапия ALPS в большинстве случаев начинается при развитии аутоиммунных цитопений. Для этого используют глюкокортикостероиды и химиотерапевтические препараты.
Спленэктомия дает лишь временное гематологическое улучшение, не предотвращает возможности последующего развития иммунных цитопений, повышает риск тяжелых инфекционных осложнений.
Роль трансплантации стволовых гемопоэтических клеток при ALPS в данный момент находится на этапе изучения.
Прогноз
Прогноз неблагоприятный. У пациентов с врожденными формами ALPS риск возникновения неходжкинской лимфомы в 17 раз выше, чем в популяции, а лимфомы Ходжкина – в 51 раз. Также возможна трансформация в аутоиммунную патологию, прежде всего, системную красную волчанку.
Клинический случай
Мальчик, 4 года 7 месяцев. Жалобы на прогрессирующее увеличение лимфатических узлов, селезенки в течении двух лет, частые отиты, пневмонии.
Анамнез жизни: до двух лет – без особенностей, после двухлетнего возраста – 5 отитов, 6 эпизодов бронхита с бронхообструктивным синдромом, 2 пневмонии.
Вакцинальный анамнез – привит не в полном объеме (имеет БЦЖ, 2 аКДС, 2 полио, 2 хиб, 1 гепатит В). Далее вакцинация не проводилась в связи с появлением описанных выше жалоб.
Ранее пациент был госпитализирован в детский гематологический стационар по месту жительства для обследования. Развернутый анализ крови, биохимические показатели, вирусологический скрининг в норме. Компьютерная томография (КТ) – признаки лимфопролиферативного процесса во всех группах лимфатических узлов. Выполнена костно-мозговая пункция – вариант нормы. Проведена биопсия пахового лимфатического узла с гистологическим и иммуногистохимическим исследованием биоптата – смешанная (кортикальная и паракортикальная) гиперплазия лимфатического узла. Пациент выписан из отделения с диагнозом «Лимфаденопатия неуточненная» под амбулаторное наблюдение педиатра, детского хирурга, детского гематолога.
На момент осмотра: общее состояние ребенка не нарушено. Лимфопролиферативный синдром представлен гиперплазией лимфатических узлов (подчелюстные, передне- и заднешейные, над- и подключичные, подмышечные, паховые размерами от 1*2 см до 3*3 см; лимфатические узлы всех доступных пальпации групп подвижны, безболезненны), гепатомегалией (+1,5 см из-под края реберной дуги) и спленомегалией (+4 см из-под края реберной дуги).
Ребенок дообследован.
Развернутый анализ крови – нормохромная нормоцитарная норморегенераторная анемия легкой степени (гемоглобин 106 г\л).
Биохимический анализ крови – незначительное повышение уровня альфа-амилазы, умеренное повышение уровня ЛДГ.
УЗИ ОБП – увеличение печени, спленомегалия с лимфаденопатией.
КТ с в\в усилением – признаки шейной, аксиллярной, внутригрудной, внутрибрюшной и тазовой лимфаденопатии (лимфатические узлы шеи образуют конгломераты общими условными размерами 34*22 мм, над- и подключичные лимфоузлы билатерально до 1 см, подмышечные – до 20*10 мм, внутригрудные – до 12 мм, внутрибрюшные и тазовые – до 20 мм). Гепато-, спленомегалия (краниокаудальный размер правой доли печени до 120 мм, селезенка увеличена до 134*57 мм).
На основании наличия у пациента хронической незлокачественной неинфекционной генерализованной лимфаденопатии, а также гепато-, спленомегалии заподозрен аутоиммунный лимфопролиферативный синдром. Пациент направлен на дообследование и консультацию детского иммунолога.
Выполнено:
Иммунофенотипирование лимфоцитов периферической крови – выявлено повышение двойных негативных лимфоцитов (CD4-CD8-) до 31,5%.
Уровень витамина В12 – значительно повышен – 1920 пг\мл (норма 191–663 пг\мл).
Определение сывороточных иммуноглобулинов G, M, A – гипергаммаглобулинемия.
Генетическое обследование – выявлена гетерозиготная мутация в гене FAS.
Пациенту был выставлен окончательный диагноз – аутоиммунный лимфопролиферативный синдром, обусловленный гетерозиготной мутацией в гене FAS.
На данный момент специфическая терапия не показана.
Ребенок продолжает находиться под наблюдением детского иммунолога и гематолога. К моменту написания данной статьи пациент вакцинирован в полном объеме согласно Календаря прививок, а также дополнительно против гепатита А, пневмококковой инфекции, менингококковой инфекции (серогруппы А, С, Y, W-135), гриппа. В ближайшее время планируется проведение вакцинации против менингококковой инфекции группы В за границей.

коментарів