Ранній вік настання менархе і ризик гестаційного діабету
Результати когортного дослідження здорових малюків
Cкорочений виклад Зореслави Городенчук*
* Early age at menarche and gestational diabetes mellitus risk: Results from the Healthy Baby Cohort study. H. Lia, L Shena, L. Songa, B. Liua, X. Zhengb, S. Xub, Y. Wanga. а – Департамент з охорони здоров'я матері і дитини, школи охорони здоров'я, медичний коледж Тунцзі, Університет наук і технологій Хуажонг, Ханконг Роуд 13, 430030 Хубей, Ухань, Китай. b – Головна лабораторія охорони навколишнього середовища та охорони здоров'я,
Міністерство освіти і Міністерство охорони навколишнього середовища та Державна лабораторія гігієни навколишнього середовища
Скорочення: ГЦД, гестаційний цукровий діабет; КЗМ, когорта здорових малюків; IADPSG, Міжнародна асоціація груп дослідження діабету і вагітності; ОГТТ, оральний глюкозотолерантний тест; ГЗСГ, глобулін, який зв'язує статеві гормони.
Детальніше
Зовнішній поворот плода на голівку і зниження числа випадків тазового передлежання
Cкорочений виклад Володимира Голяновського
Пропонуємо познайомитись з науково-практичним керівництвом RCOG з практики зовнішнього повороту плода на голівку у разі тазового передлежання. Метою даного керівництва є опис і узагальнення найбільш переконливих доказів щодо методів запобігання не-головному передлежанню плода під час пологів і пов'язаним з цим виконанням кесаревого розтину з притаманними йому ускладненнями. Дане керівництво опубліковано на зміну керівництву, яке вперше було опубліковане 2006 року
Детальніше
Що таке Cocoonababy®?
Новонароджені малюки сплять до 20 годин на добу, ось чому так важливо створити для них сприятливу та безпечну атмосферу для здорового сну. Cocoonababy® від Red Castle – це ергономічний кокон, який можна використовувати в будь-який час дня чи ночі задля комфорту та благополуччя малюка. Завдяки кокону Cocoonababy® дитина знаходиться у згрупованій позі, яка, нагадуючи внутрішньоутробну, заспокоює малюка та забезпечує плавний період адаптації у перші місяці після народження. Розслаблений малюк краще засинає та гармонійно проходить фази сну.

Оформити замовлення на Cocoona baby можна на сайті дилера в Україні www.lebebe-boutique.com чи за телефоном (050) 709-60-84
Якi переваги використання Cocoonababy?
Поза в Cocoonababy дозволяє дитині плавно пройти перехідний період між внутрішньоутробним і зовнішнім світом. Малюк відчуває певне обмежене середовище, яке, однак, не обмежує його рухів, – саме ці відчуття він переніс, коли знаходився у маминому животику.
Cocoonababy дозволяє мінімізувати найбільш поширені проблеми, з якими стикається малюк в перші місяці після народження:
- Покращує якість та тривалість сну;
- Усуває пробудження від різкого здригання (рефлекс Моро);
- Знижує проблеми шлунково-стравохідного рефлюкса;
- Знижує ризик виникнення синдрому сплюснення голови (плагіоцефалії);
- Забезпечує кращий розвиток моторики, зорово-моторної координації;
- Покращує контакт між дитиною та її оточенням.


Хто розробив Cocoonababy?
Форма кокона розроблена за ініціативою Даніеля Сельдуччі – кінезіотерапевта, який спеціалізується у галузі педіатрії, спільно з докторами Палі, Фабр-Грене і командою спеціалістів Північного Госпіталю Марселя (Франція).
Cocoonababy є результатом 10-річних спостережень і досліджень лікарів в області позиціонування новонароджених. Сьогодні кокон використовується у багатьох госпіталях, пологових будинках, а також практикуючими спеціалістами (педіатрами, акушерми, остеопатами і т. д.).
Вiдгуки батькiв
Кокон був з нами з самого народження у пологовій палаті. Спочатку ми використовували його постійно, переносячи малюка з собою з кімнати в кімнату. Це дуже зручно, оскільки завжди є можливість бути поряд з малюком, не хвилюватись за його стан та відповідати на його бажання. Ну і, звичайно, для молодих батьків дуже важливо постійно бачити малюка для встановлення контакту, а в моєму випадку – для особистого спокою. Також нам сподобалось те, що завдяки чохлу з ручками, є можливість брати кокон з собою в гості. Так у малюка завжди є своє звичне ліжечко та середовище, йому комфортно та затишно. З перших годин перебування вдома ми з чоловіком помітили, що малюку подобається перебувати в коконі, його не турбують коліки, він зразу заспокоюється і засинає.
Таня, мама Максима
Вiдгуки професiоналiв:
В коконі дітям дуже комфортно. В ньому новонароджений перебуває в фізіологічному положенні, як плід під час внутрішньоутробного розвитку.
Новонароджений відчуває дискомфорт, коли лежить на великому ліжку, тому кокон з його обмеженим простором – найкраще ложе для нього. В пологових будинках малюкам в коконах зручніше проводити фототерапію. Під спеціальними лампами діткам доводиться лежати не одну годину. Тому фізіологічна форма кокона є просто порятунком. Немовлята лежать в природній позі, їм зручно і тепло. Фототерапія проходить більш успішно.
Лікар-педіатр Волинської обласної дитячої лікарні Грабовська Ірина Михайлівна
У кокона багато переваг: можливість корекції гіпертонусу, асиметрії черепа, регургітації…
Поза, в якій малюк знаходиться в коконі, гарантує безпеку крижово-клубового зчленування, завдяки згрупованому положенню, яке нагадує внутрішньоутробне; немовля приймає оптимальну позу тіла і голови для здорового контакту із оточенням; можливість вільної жестикуляції для малюка і візуального контролю над всім, що відбувається зі сторони дорослих
На даний момент, не існує аналога Cocoonababy, який дозволяє забезпечити зниження гіпертонусу і гіперактивності у дитини при важких пологах чи ускладненій вагітності.
Сімейний психотерапевт, неонатолог Екс-Ан-Прованс Марі Шміт
Правильне позицiонування, чому це так важливо?
Як правильно розмiстити малюка?
Завдяки спеціальному валику, який розташовується під чохлом, Ви завжди зможете розмістити малюка у зручній і правильній позі.


Iнновацiйний та високотехнологiчний продукт
На відміну від зберігаючих форму пінополіуретану чи мікрокульок, Cocoonababy створений з піноматеріалу високої резистентності, який не обмежує свободу рухів. Гарантована якість та збереження форми після тривалого використання.
Вигляд пiноматерiалу в розрiзi
Перший, більш м'який шар, – для ніжної підтримки голови дитини. Нижній шар, щільніший, – для зручності та комфорту.
Знак якості Certipur гарантує виробництво піноматеріалу згідно найвищих стандартів гігієни та безпеки завдяки сировині, яка не забруднює навколишнє середовище. Вся тканина відповідає стандартам Oekotex 100.
Комплексний продукт:
Верхній чохол, виготовлений із фірмової бавовни Fleur de Coton; пояс для більшої безпеки; захисний чохол із тканини, яка не промокає та дихає; зручний чохол з ручками для перевезень.
Для гарантії комфорту та безпеки малюка експерти підібрали високотехнологічні та безпечні матеріали.

Якi аксесуари можна використовувати з Cocoonababy?
- Знімний чохол.
- Знімний захисний чохол
- Знімний пояс
- Ковдра для кокона Cocoonababy. Ковдри з ніжної бавовни Fleur de Coton повторює форму кокона і защіпається на грудях за допомогою липучки. Мамі не варто хвилюватися, що малюк розкриється чи натягне ковдру на обличчя.
Технiчнi характеристики
- Використовується з народження (з 2,8 кг) до того часу, коли малюк, знаходячись в коконі, почне перевертатися чи змінювати положення тіла (приблизно 4 місяці +);
- Використовувати на підлозі, килимі чи в ліжку. Глибина від поверхні, на яку встановлюється Cocoonababy, повинна перевищувати 34 см. Не рекомендується використовувати в колисці, колясці;
- Cocoonababy інтенсивно використовується в пологових будинках. Його високоякісна основа розроблена для того, щоб зберегти первісну форму. При дбайливому використанні, дотриманні гігієни і правильному зберіганні Cocoonababy можна використовувати для декількох дітей;
- Розмір (довжина x ширина х висота) 69х40х19 см. Вага: 1 кг.
- Догляд: верхній чохол і знімний поясок допускається прати у пральній машинці;
- Склад: відповідає стандартам безпеки і стандартам аксесуарів для сну №2000-164 від 23.02.2000;
- Гарантія: 6 місяців;
- Склад: Основа – PU; Верхній чохол – 100% бавовна; Захисний чохол – 100% Tencel. Мембрана – 100% PU; Змінний поясок – 100% бавовна.
Нейрональний цероїдний ліпофусциноз
Під терміном «нейрональний цероїдний ліпофусциноз» (NCL) розуміють найбільш розповсюджену в дитячому віці групу клінічно та генетично гетерогенних нейродегенеративних захворювань, що характеризуються внутрішньоклітинним накопиченням автофлюоресцентних ліпопігментів у різних ультраструктурних локусах та клінічно проявляються втратою зору, швидко прогресуючою деменцією, епілептичними нападами, моторними розладами і ранньою смертю
У літературі захворювання даної групи описані як хвороба Баттена, хвороба Перрі, хвороба Шпільмейєра–Шегрена, хвороба Більшовського, хвороба Куфтса, хвороба Сантавуорі–Халтіа.
Перша згадка про нейрональний ліпоїдний ліпофусциноз з’явилась в 1826 році і належить Stengel, який описав перших 4 пацієнтів з Норвегії. Перший клініко-патологічний опис (1903) належить Batten. Він же охарактеризував NCL, як «сімейну нервово-м'язову дегенерацію». Також Batten був першим, хто диференціював NCL з хворобою Тея–Сакса у 1914 році. Приблизно в цей же час Vogt, Spielmeyer, Bielschwsky і Kufs також описали пацієнтів старшого віку з симптомами, схожими на описані Batten. Однак повноцінне вивчення захворювань даної групи почалося з 1980-х, з розвитком біохімії і молекулярної генетики.
Епідеміологія
Як вже було зазначено, дана група дуже гетерогенна за своїм клінічними, патофізіологічними та генетичними характеристиками, тому досить важко говорити про частоту даного захворювання. Однак, згідно з наявними даними, в США налічується близько 25000 сімей з встановленим діагнозом NCL.
Міжнародні дані: у фінській популяції NCL1 зустрічається з частотою 1:20 000, та 1 особа із 70 є носієм патологічного гену; поширеність NCL2 в світі — 0,6–0,7 на мільйон із захворюваністю на рівні 0,46 на 100 000 живих новонароджених; NCL3 – друга за частотою форма NCL в світі (7 випадків на 100 000 живих новонароджених в Ісландії). Найбільш висока поширеність NCL відзначається у скандинавських країнах, особливо в Фінляндії.
Патофізіологія
При всіх формах NCL відбувається накопичення у лізосомах клітин автофлюоресцентного ліпопігменту, який складається з білків сапозинів А і D і/або субодиниці з мітохондріальної АТФ-синтази.
Матеріал, що накопичується, характеризується автофлюоресценцією в зелено-жовтому спектрі при збудженні світлом довжиною від 340 до 360 нм.
При гістохімічному фарбуванні матеріал дає позитивну реакцію на кислу фосфатазу і забарвлюється суданом чорним В, що свідчить про присутність фосфоліпідів. Він також є ШИК-позитивним, що може вказувати на високий вміст вуглеводів. Більшість з цих властивостей ідентична з властивостями так званих пігментів «зношування», або «старіння», яких ще називають ліпофусцинами або цероїдами. При електронній мікроскопії нейрональних і екстранейрональних тканин виявляють цитосоми, заповнені агрегатами характерної морфології, що описуються, як «криволінійні» профілі, за типом «відбитків пальців», «прямолінійні» профілі, гранулярні осміофільні відкладення.
Ліпофусцин – дрібний, гранулярний, золотисто-коричневий пігмент, що складається з фосфоліпідів і білків. Ліпофусцин накопичується в організмі тварин і людини в нормі, у міру їх зростання і старіння. Залежно від віку, умов утворення і локалізації розрізняється за гістохімічними і ультраструктурними характеристиками, спектрами флуоресценції і поглинання, розчинності в органічних розчинниках.
Цероїд – ліпопігмент, який утворюється в макрофагах шляхом гетерофагії при резорбції ліпідів. Містить нерозчинні в спирті ремнанти окислених ЛПНЩ, які здатні витримувати лізосомальний гідроліз в макрофагах. Механізм утворення цероїду можна уявити наступним чином: після ендоцитозу ліпідів з утворенням ліпофагосом, відбувається їх часткове перетравлювання у вторинних лізосомах з утворенням третинних лізосом, або телолізосом, які містять речовину, що зветься цероїдом.
Спочатку класифікація NCL ґрунтувалася на віці початку маніфестації та симптоматиці захворювань і налічувала 4 типи:
- Тип I – рання дитяча форма.
- Тип II – пізня дитяча форма. Хвороба Більшовського–Янського.
- Тип III – підліткова форма. Хвороба Баттена–Шпільмеєра–Фогта.
- Тип IV – пізня форма. Хвороба Куфса.
На теперішній момент класифікація доповнена даними генетичних досліджень і набула наступного вигляду (табл. 1).
Всі форми захворювання мають аутосомно-рецесивний тип спадкування.
Характеристика окремих типів NCL
NCL 1, хвороба Сантавуорі–Халтіа, інфантильний ліпофусциноз.
Захворювання пов'язане з мутацією гена 1р32, відповідального за синтез пальмітоїл-протеїн тіроестерази-1 (PPT-1), яка видаляє ацильні групи цистеїну на ліпопротеїдах ЕПС.
Розрізняють наступні форми захворювання:
- Класична інфантильна
1968 – Herbert et al. описав прогресуючу енцефалопатію у дитини з фінської сім'ї.
1973 – Santavuori et Haltia описали основні клінічні та морфологічні особливості даної форми – мікроцефалія, гіпотонія, гіперзбудливість, когнітивні розлади, зорові порушення, атаксія, екстрапірамідні порушення, спастичність, міоклонус, втрата моторних і соціальних навичок до 2 років, смерть до 6–13 років.
- Пізня інфантильна
1979 – Becker в Німеччині описав дитину, у якого зорові і ментальні розлади почалися до 3 років, з попередніми міоклонічними нападами. Відмічалось прогресуюче зниження когнітивних функцій, клінічно форма нагадує NCL2, смерть до 10–13 років.
- Ювенільна
1995 – опис належить Philippart et al. і Hofman and Taschner. Спостерігалася втрата зору і здатності до придбання нових навичок до 5–7 років, клінічно схожа з NCL3, але з пізнім початком епілептичних нападів і більш ранніми моторними порушеннями
- Доросла форма
2001 – Van Diggelen описав 2 сестер, з початком NCL1 в дорослому віці і встановленими мутаціями гена РРТ1.
2007 – Ramadan et al. був описаний випадок NCL1 у 24-річної жінки. Виявлено маніфестацію до 30 років, психічні порушення з прогресуючою когнітивною дисфункцією, атаксію, паркінсонізм, атрофію зорового нерва; пацієнти з цією формою захворювання доживають до 50 років.
В цілому характеристика даного типу відображена в табл. 2.
NCL 2, хвороба Більшовського–Янського, пізній інфантильний NCL.
Мутація гена 11р15.5, відповідального за синтез лізосомльного ферменту трипептид-пептідази-1, що відщеплює трипептиди з N-поліпептидного кінця.
Розрізняють наступні форми:
- Пізня інфантильна
1926 – Hassin належать перші відомості про дану форму захворювання.
1957 – Seitelberger та ін. визначають в літературі 28 випадків. Маніфестація у віці 2–4 років, епілептичні напади, зниження когнітивних функцій, атаксія, міоклонус, екстрапірамідні розлади, повна втрата зору до 4–6 років, смерть у другій декаді життя або раніше.
- Ювенільна
1977 – Дана форма була вперше виділена Andermann та ін. Йому ж належить опис 17 випадків захворювання. Маніфестація у 6–8 років, прогресуюче зниження когнітивної функції, судоми, атаксія, моторні порушення, різні порушення зору, деякі пацієнти доживають до 40 років.
Загальна характеристика подана у табл. 3.
NCL 3, хвороба Баттена, хвороба Вогта–Шпільмейєра, хвороба Шпільмейєра–Шагрена.
Мутація гена 16р12.1, що кодує білок, який складається з 438 амінокислот та є частиною мітохондріальної мембрани. Найбільш частим видом мутації даного гена є 1.02-kb делеція.
Розрізняють:
- Ювенільну форму
1904 – Баттен описав пацієнтів з даним захворюванням та визначив прогресуюче зниження зору з 4–7 років, що приводить до повної втрати зору у 10 років, дизартрію, зниження когнітивних функцій, епілептичні напади, психічні порушення у вигляді порушення соціальної адаптації та ідентифікації, зниження уваги, підвищену агресію, паркінсонізм, міоклонус, розлади сну, мозочкові розлади та розлади екстрапірамідної системи.
Загальна характеристика подана у табл. 4.
NCL 4, хвороба Куфса, NCL дорослих.
Мутація гена NCL4. Маніфестує даний тип, як правило, у віці 30 років, проте є дані, згідно з якими можлива маніфестація і у віці 11 років.
1925 – Куфс вперше описав випадок дорослого NCL, з проявом захворювання у 26 років і смертю пацієнта в 34 роки.
Виділяють два типи даного захворювання:
- тип А: відзначаються прогресуючі міоклонічні епілептичні напади, деменція, атаксія, пірамідні і екстрапірамідні розлади.
- тип В: деменція, моторні порушення, атаксія, маніфестація можлива і після 50 років (табл. 5).
NCL 5, пізній інфантильний, фінський варіант.
Мутація гена 13q21.1-q32,що кодує трансмембранний білок, який складається з 407 амінокислот.
Перший опис хвороби даний Santavuori у 1982 р. Він повідомив про 18 фінських сімей, в яких були хворі на NCL. Маніфестація у віці 4–7 років, клінічно хвороба нагадує NCL2, але протікання більш повільне, смерть настає у другій-третій декаді життя.
Загальна характеристика подана у табл. 6.
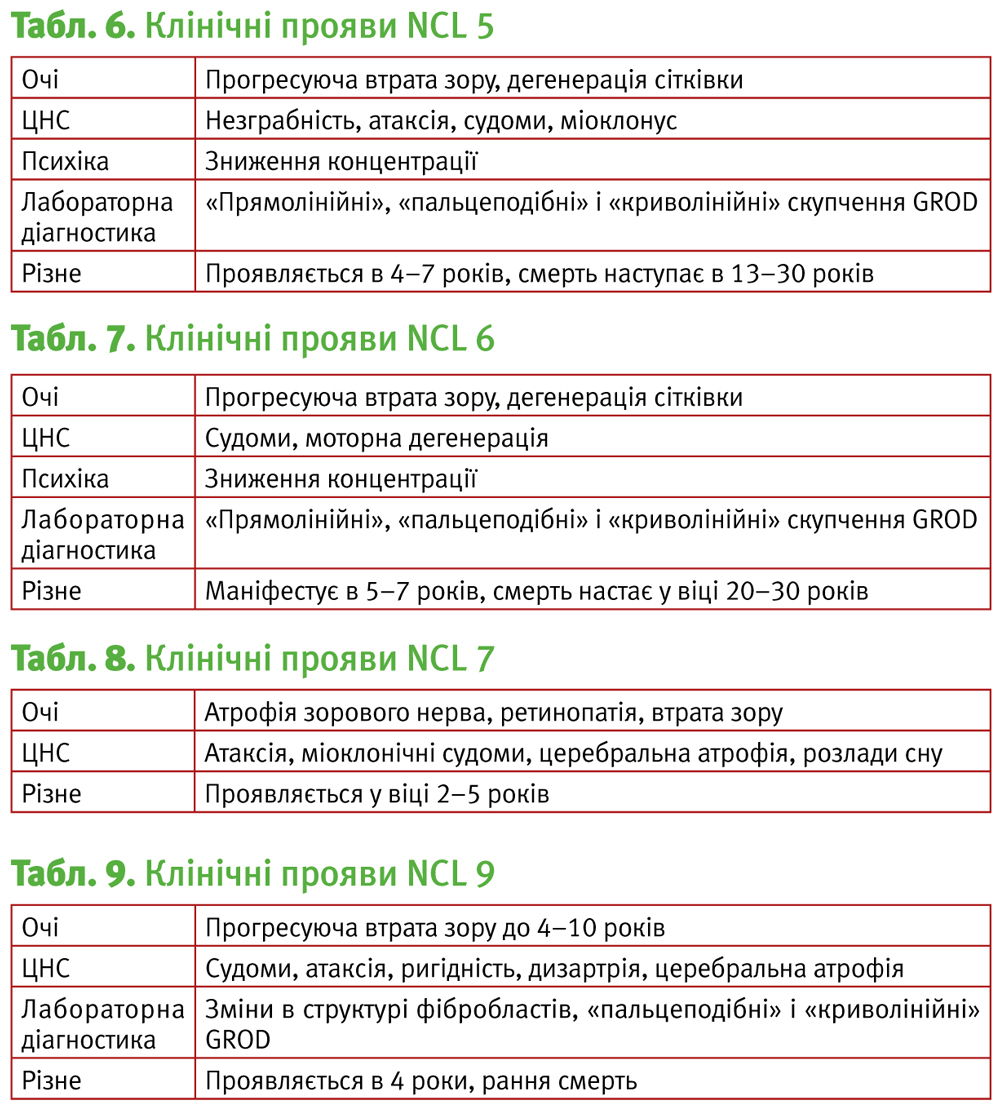
NCL 6, пізня інфантильна форма, чеський або індійський варіант.
Мутація гена 15q21-q23, що кодує мембранний білок, який складається з 311 амінокислот.
1997 – Sharp et al. описують 2 сім'ї із захворюванням, що гістологічно подібне до NCL2. Прояви хвороби, як правило, починаються від 18 місяців до 8 років у вигляді зниження або втрати зору, судом, втрати моторних навичок до 4–10 років; смерть настає у другій-третій декаді життя. Варіант захворювання клінічно схожий з NCL2 (табл. 7).
NCL 7, турецький варіант.
Захворювання викликане мутацією в гені 4q28.1-q28.2, що кодує лізосомальний мембранний переносник.
1999 – Wheller et al. описує пацієнтів з так званим турецьким варіантом NCL.
2005 – Ranta вивчає 7 турецьких сімей з даним захворюванням. Захворювання характеризується атрофією зорового нерва, атаксією, міоклонічними судомами, церебральною і мозочковою атрофією, розладами сну (табл. 8).
NCL 8, пізній інфантильний варіант, «північна епілепсія».
Мутація гена 8pter-p22, що кодує трансмембранний білок, який складається з 286 амінокислот.
1999 р. – Wheeller повідомляє про виявлення 6 пацієнтів з відомим для NCL фенотипом, але не встановленими локусом мутації.
Для NCL8 визначено 5 різних мутацій, але місценс-мутація (R24G), призводить до прогресуючої епілепсії з затримкою розумового розвитку (PERM).
Відповідно розрізняють:
- турецький варіант пізнього інфантильного NCL – проявляється у 3–7 років, прогресуюча втрата зору, судоми, інтелектуальні порушення, дизартрія, міоклонус, атаксія.
- «північну» епілепсію – епілептичні напади у 5–10 років, повільне прогресуюче зниження розумового розвитку, можливе зниження зору, деякі пацієнти доживають до 60 років.
NCL 9.
Даний тип описаний у 2004 році Schulz на 2 сестрах з Сербії та братах з Німеччини. У сестер відзначалося зниження зору, прогресуюча атаксія і судоми у 4 роки. До 10 років була втрачена здатність до самостійної ходьби. Подібна симптоматика спостерігалася і у двох братів. Зниження когнітивних функцій почалося з 6 років, атаксія і ригідність відзначалися з 9 років. Молодший із братів помер у 15 років від пневмонії,а старший, який страждав згодом дисфагією і галюцинаціями, помер у віці 19 років. Імовірно, що ген, мутація якого викликає захворювання, кодує білок, що бере участь у регуляції активності дигідроцерамідсинтетази (табл. 9).
NCL 10. Вроджений, катепсин-D-дефіцитний.
Мутація гена 11p15.5, що кодує катепсин D. Перші згадки відносяться до 1941 р. Norman та Wood, а також до 1954 р. Brown. Дана форма описана в 2006 році Steinfeld у групі із 25 дітей з не ідентифікованим NCL-подібним захворюванням. Характеризується наступними ознаками: мікроцефалія, прогресуюча втрата зору, епілептичні напади, епілептичний статус, атаксія, спастичність, ригідність, церебральна атрофія.
Діагностика
У діагностиці NCL використовують дані електрофізіологічних та гістологічних досліджень, а також різних методів візуалізації.
Магнітно-резонансна томографія (МРТ): NCL1 – слабка церебральна атрофія, прогресуюча після 4 років, зниження T2-сигналу в області таламуса, прогресуюче зниження контурованості білої речовини на T2, атрофія мозочка з 3 років, витончення поясної звивини. CLN2 – прогресуюча, особливо субтенторіальна, атрофія мозку. CLN3 – церебральна атрофія, зазвичай після 15 років. NCL6 – поширена церебральна і мозочкова атрофія.
Позитронно-емісійна томографія (ПЕТ): NCL2 – генералізований гіпометаболізм. CLN3 – гіпометаболізм в області шпорної борозни.
Магнітно-резонансна спектроскопія (МРС): NCL1 – майже повна втрата N-ацетиласпартату, редукція креатин- і холін-вмісних сполук, зростання міоінозитолу, зростання вмісту лактату в сірій і білій речовині.
ЕЕГ: NCL1 – втрата атенуації при відкриванні очей, згладжування «веретен сну». NCL2 – потиличні спайки 1-2 Hz зі світловим подразником.NCL3 – спайки і повільно-хвильові комплекси.
Електроретинограма: NCL1 (інфантильна форма) – неможливо записати до 3 років. NCL1 (ювенільна форма) – неможливо записати. NCL2 – неможливо інтерпретувати, в подальшому зникає. NCL3 – ранні аномалії при реєстрації.
Зорові викликані потенціали: NCL1 – неможливо записати з 4 років. NCL2 – відзначаються аномалії, але інтенсивність сигналу знижується на фінальних стадіях. NCL3 – ранні аномалії при реєстрації.
Гістологічне дослідження. NCL1 – майже повне зникнення кортикальних нейронів. NCL3 – вакуолізовані лімфоцити, селективний некроз клітин-сателітів 2 і 3 шару кори, і втрата пірамідних клітин 5 шару. NCL5 – дегенерація нейронів неокортексу і мозочка, дегенерація 3 і 5 шарів кори, багатоядерні нейрони в 3 шарі кори. NCL6 – втрата нейронів 5 шару, зниження кількості клітин Пуркіньє, відсутність SCMAS в печінці, наднирковій і підшлунковій залозах. NCL8 – дегенерація клітин 5 шару і нейронів гіпокампу, зниження SCMAS в клітинах Пуркіньє.
Остаточний діагноз верифікується за допомогою ДНК-тестування з визначенням мутантного гена.
Диференціальна діагностика
Дифдіагностика проводиться з абсансною епілепсією, комплексними парціальними судомами, допамін-чутливою дистонією, Epilepsia Partialis Continua, атаксією Фрідрейха, хворобою Халлевордена–Шпатца, хворобою Гантінгтона, Retinitis pigmentosa, синдромом Ретта, метаболічними та інфекційними ушкодженнями ЦНС.
Лікування
Специфічної патогенетичної терапії не розроблено. Лікування симптоматичне (антиконвульсанти), використання антиоксидантів безрезультатні, проводилися спроби пересадки кісткового мозку, але ця методика не виявилася дієвою.
У якості ілюстрації наводимо власне спостереження випадку нейронального цероїдного ліпофусцинозу ІІ типу у дитини.
Хлопчик Д., 5 років, зі скаргами на напади судом, які почастішали у динаміці.
Анамнез: друга вагітність, перші пологи. Перша вагітність закінчилась викиднем. Пологи – кесарів розтин на 40-му тижні, вага при народженні 4050 г. Постнатальний період в нормі. Почав стояти у 7–8 місяців, ходив самостійно в 10 місяців. Мовні навички: перші слова у віці одного року.
В 3 роки у дитини почалися судомні напади різного типу, включаючи генералізовані міоклонічні судоми. Хода дитини стала нестабільною. Спостерігалося легке відставання в мовному розвитку. Лікувався у міському неврологічному відділенні. Було встановлено діагноз: Злоякісна міоклонічна епілепсія. Аутизм. Призначено лікування комбінацією антиконвульсантів, яке не принесло бажаного ефекту. З січня 2015 р. (4 роки) хлопчик не вимовляє слів, поведінка аутична. Спостерігалась мовна регресія протягом 9 місяців. В останні 6 місяців спостерігався моторний і когнітивний регрес. З квітня 2015 р. – посилення судом і регресія, в основному, мовних навичок. Моторно — наросла м’язова слабкість, іноді міг йти сам, іноді потребував допомоги. Також відмічалася регресія контролю над природними відправленнями.
Хлопчик був спрямований на обстеження у Центр орфанних захворювань, де встановлено діагноз: Епісиндром. Затримка мовного розвитку. Було рекомендовано консультування і обстеження в медичному центрі «Асафа-Рофе» (Ізраїль) на предмет діагностики спадкових нейродегенеративних захворювань.
У квітні 2015 р. в медичному центрі «Асафа-Рофе» були проведені наступні дослідження:
Лабораторні аналізи: електроліти та функції печінки в нормі, функції нирок у нормі, СРБ нульовий. Вальпроєва кислота 34,4. ОАК: лейкоцити 8,6 гемоглобін 11,7, тромбоцити в нормі.
Метаболічні дослідження: вміст у крові амінокислот, карнітину, ацилкарнітинів, аміаку, піровиноградної кислоти – без відхилень від норми. Рівень лактату трохи підвищений — 25 (норма до 18), сеча на сечову кислоту — в нормі.
ЕЕГ: полівогнищева епілептична активність (ліва лобова частка, права тім'яна частка). Короткі (1–5 секунд) спалахи генералізованих тета-хвиль високої напруги зі змішаними піками.
МРТ мозку: гіпоплазія мозочка, зменшення об'єму супратенторіальної та інфратенторіальної тканини мозку, генералізована атрофія паренхіми, дилатація шлуночків.
Генетична діагностика: проведено генетичний чіп – без хромосомних змін, пов'язаних з високою ймовірністю з відомим генетичним або неврологічним синдромом.
Секвенування екзома: виявлено в екзоні 6 гетерозиготний патогенний варіант гену ТТР-1, с.622С>Т, також у екзоні 7 гетерозиготний варіант гену ТТР-1, с.833А>С, що свідчить про підтвердження діагнозу «Нейрональний цероїдний ліпофусциноз, тип 2».
Клінічний діагноз: Нейрональний цероїдний ліпофусциноз, тип 2. Генералізована епілепсія з міоклонічним компонентом.
Рекомендовано:
- Почати терапію фелбаматом в дозі 150 мг двічі на день;
- Почати терапію клобазамом в дозі 10 мг двічі на день;
- ЕЕГ через три місяці;
- Контрольна МРТ через рік для спостереження;
Призначена терапія не принесла бажаного результату. Періодично виникали судоми. З вересня 2016 року, судоми відзначались покілька разів на тиждень. Протягом останніх трьох днів перед госпіталізацією було 3–5 судом в день. 21.09.16 стан погіршився, додому була викликана бригада швидкої допомоги, вдома введено сибазон. Дитину з епістатусом було госпіталізовано у ВАІТ ДМКЛ №6.
При огляді у ВАІТ: стан дитини тяжкий, обумовлений неврологічною симптоматикою. t – 36,5ОС; ЧСС – 90/хв.; ЧД – 26/хв.; АТ – 110/60 мм рт. ст.; SpO2 – 99%. Свідомість – медикаментозний сон. Менінгеальні симптоми негативні. Зіниці D=S, обличчя симетричне. Плаваючі рухи очних яблук. Короткочасні тонічні напруження кінцівок. Шкірні покриви бліді, теплі, висипань не спостерігається. Тургор м’яких тканин та еластичність шкіри достатні. Видимі слизові порожнини рота рожеві, помірної вологості. Задишки та видимих гіпоксичних розладів при диханні атмосферним повітрям немає.
Аускультативно: в легенях дихання жорстке, проводиться рівномірно в усі відділи. Перкуторно над легенями визначається легеневий звук з обох боків. Тони серця приглушені, ритмічні. Видимих набряків немає. Живіт м’який, доступний глибокій пальпації. Печінка +1,0 см. Біля краю реберної дуги. Селезінка не пальпується. Фізіологічних відправлень при огляді не було.
Лабораторні дослідження: загальний аналіз крові, загальний аналіз сечі, нирково-печінковий комплекс, електроліти, глюкоза – у межах вікової норми.
ЕЕГ: полівогнищева епілептична активність.
Консультація невролога: нейрональний цероїдний ліпофусциноз. Симптоматична епілепсія. Груба затримка психо-моторного розвитку. Стан після епілептичного нападу.
Клінічний діагноз: Нейрональний цероїдний ліпофусциноз, ІІ тип. Симптоматична епілепсія. Епістатус.
У відділенні реанімації отримував наступну терапію: препарати вальпроєвої кислоти, карбамазепін, клобазам. Сибазон за показаннями.
Через 3 дні стан дитини покращився. Судомний синдром був купіруваний. Дитина у стабільному стані переведена для подальшого лікування у відділенні неврології.
Висновки
Сьогодні перелік орфанних захворювань стає все об'ємнішим, а можливості їх діагностики – доступнішими. У даному випадку наявність у дитини поєднання епілептичних нападів із ознаками нейродегенеративного захворювання стало підставою для пошуку генетичної патології. Уточнення діагнозу дозволяє визначити прогноз та в ряді випадків оптимізувати терапію. Тому ми вважаємо за доцільне нагадати основні показання для спрямування пацієнта з неврологічними порушеннями у медико-генетичні центри та центри метаболічних захворювань:
- епілептичні судоми, що важко піддаються належній медикаментозній корекції;
- ознаки нейродегенеративних захворювань центральної нервової системи у поєднанні з захворюваннями органів зору, слуху, атаксіями та моторними порушеннями;
- МРТ-ознаки лейкодистрофії нез’ясованої етіології;
- невпинна втрата психомоторних навичок, набутих раніше;
- моторні порушення у поєднанні з мальформаціями Денді–Уокера та іншими гіпоплазіями мозочка;
- міопатичні синдроми;
- полінейропатії, не пов’язані з інфекційно-алергічною природою;
- розумова відсталість, аутична поведінка та інші порушення поведінки, поєднані з набутими порушеннями зору, слуху та моторними порушеннями.

Захід зустрічає схід: педіатрична практика в системі громадського здоров’я
Під такою назвою 31 березня 2017 року в Києві в рамках Х Національного конгресу «Людина та ліки» – Україна 2017 пройшла науково-практична конференція, організована Громадською організацією «Українська Академія Педіатрії».
«Українська Академія педіатрії» (УАП) – організація, створена нещодавно, попри це, про неї аж ніяк не можна сказати «маловідома», адже свою діяльність УАП розпочала дуже активно і вже встигла провести чимало освітніх заходів у різних містах України. Адже, як зауважив віце-президент УАП, лікар-педіатр, к. мед. н. Ярема Возниця, «головне завдання цієї організації – вчити педіатрів і сімейних лікарів новим технологіям задля досягнення кінцевої мети – підвищення здоров’я дітей, а отже, і громадського здоров’я. Тому ми запровадили і проводимо курс лекцій, майстер-класів і практичних занять по всіх областях України».
Президент ГО «Українська Академія Педіатрії», дитячий гематолог, д. мед. н., проф. каф. педіатрії і неонатології факультету післядипломної освіти ЛНМУ ім. Данила Галицького Леонід Дубей також відзначив:
«Своєю діяльністю УАП прагне розширити зв’язки між медичними спільнотами України та зарубіжних держав, сприяти впровадженню в Україні міжнародних стандартів надання медико-соціальної допомоги дітям, реалізації захисту їхніх прав і свобод. Важливою подією як для європейської спільноти, так і для України є рішення Генеральної Асамблеї Європейської Академії Педіатрії 31 січня 2016 року у Брюсселі щодо набуття статусу асоційованого члена Громадською організацією «Українська Академія Педіатрії», яка стане достойним представником української педіатрії у Європі. Підтримка міжнародного педіатричного співтовариства, а також обмін знаннями є дуже важливими на шляху до Європейського Союзу». Професор Дубей також висловив своє переконання у тому, що саме об’єднання профільних громадських організацій та асоціацій за європейським взірцем є дуже важливим для спільного представництва у європейських структурах, а також узгодження перспектив розвитку педіатричної галузі в цілому. «Спільними зусиллями ми досягнемо більшого!», – цим гаслом Європейської Академії Педіатрії, яке тепер стало і нашим, закінчив свою вітальну промову до учасників цієї науково-практичної конференції Леонід Дубей, передавши слово для привітання в. о. міністра охорони здоров’я України Уляні Супрун.
В своєму виступі п. Уляна досить розлого зупинилася на найбільш актуальному питанні – реформі системи охорони здоров’я, що нині відбувається в країні: «Ми не вигадували велосипед, натомість в основу концепції реформи поклали досвід Великої Британії, Іспанії, Норвегії та багатьох інших країн світу, в яких система охорони здоров’я гарантує громадянам якісні і доступні медичні послуги, а лікарям – належні умови і оплату їхньої праці».
Вона також повідомила, що до Верховної Ради України подано законопроекти, прийняття яких дозволить втілювати реформи. Але працювати потрібно, особливо підкреслила в. о. міністра, у досить швидкому темпі, тож вона закликала лікарів не бути осторонь цього процесу, спонукати місцевих депутатів голосувати за ці зміни у Верховній Раді. Сьогодні першочерговими завданнями є: змінити систему фінансування медицини, закріпити педіатрію на первинній ланці шляхом внесення змін до Основ законодавства України про охорону здоров’я (МОЗ це вже зробило відповідними наказами).
«Як зміниться система охорони здоров’я, коли всі законопроекти буде проголосовано? По-перше, це концепція «Гроші йдуть за пацієнтом». Друге – це персоніфікація медичних послуг і третє – перехід на міжнародні протоколи лікування. Останнє дуже важливо, адже ми маємо на сьогодні застарілі протоколи (у педіатрії, зокрема). Звичайно, має бути перехідний період, не можна відразу перейти до міжнародних стандартів, викинувши все українське. Разом з тим, потрібно зауважити, що система може бути прописана чудово, може бути безліч протоколів тощо, але, якщо ніхто їх не виконує, то нічого не станеться. Імплементація дуже важлива. Зараз є проблема з нашими законами взагалі. Наприклад, ми маємо чудовий закон про екстрену медичну допомогу, відповідно до якого, кожна лікарня повинна мати відділення невідкладних станів, але чи вони існують?!», – риторично запитує Уляна Супрун. – Отже, імплементація всього того, що міститься в законах, залежить від вас, лікарів. Саме ви маєте втілювати зміни на місцях. Дуже важливо, щоби наші лікарі, зокрема, головні лікарі, були прогресивними і хотіли змінити систему. Адже система – це не міністерство, лікарні або асоціація. Система – це всі ми», – переконана Уляна Супрун.
Д. мед. н., директор ДУ «ІПАГ НАМНУ», президент Асоціації педіатрів України Юрій Антипкін, перш за все, подякував п. Супрун за те, що, завдяки її підтримці, вдалося відстояти сьогодні педіатрію на первинному рівні. Він також відзначив декілька аспектів, що вирізняють цю конференцію з поміж інших. Зокрема, окрім освітньої місії, важливим є те, що педіатрія тут розглядається в аспекті громадського здоров’я, адже є важливою його складовою. Не може бути здорової нації без міцного здоров’я дитячої популяції населення. Тож обговорення актуальних проблем медицини дитячого віку допоможе визначитися зі стратегічними напрямками розвитку педіатрії, головним з яких Юрій Антипкин вважає профілактику. В цьому аспекті вчений-клініцист наполягає на тому, що лікарі мають більше уваги приділяти саме збереженню здоров’я дитини, а не тільки лікуванню: «Адже, на сьогоднішній день ми маємо вражаючі результати дослідження здоров’я дітей шкільного віку, згідно яких, за час навчання, частка здорових дітей знижується, в середньому, втричі (!), що свідчить про зниження ефективності проведення профілактичних заходів», – констатує Юрій Антипкін.

Перед стартом науково-практичної частини конференції, з важливим повідомленням виступив Тимофій Бадіков, голова правління ГО «Батьки за вакцинацію». Він коротко розповів про заходи, які проводить ГО, зокрема, в рамках Українського тижня імунізації що проходив 24–30 квітня цього року, анонсував події, які мають відбутися найближчим часом, запросивши присутніх взяти в них участь, а також закликав лікарів ставати «агентами змін з вакцинації», запропонувавши заповнити невеличку анкету, що забезпечить зворотній зв’язок і дозволить розпочати співпрацю: організувати лікарю участь у тренінгу для агентів змін; отримати довідник «Відповіді на складні запитання батьків про вакцинацію», який було створено у співпраці з головним дитячим імунологом Києва Федором Лапієм, та інші корисні матеріали, які можуть допомогти лікарю в роботі. Адже запитань щодо вакцинації, зазвичай, дуже багато.
Продовжилася конференція доповіддю д. мед. н., проф. кафедри педіатрії №2 НМАПО ім. П. Л.Шупика Марини Маменко «Йодний дефіцит та його наслідки: як можна вирішити національну проблему?».
Вона зупинилася на основних моментах, зокрема зазначивши, що йододефіцит (ЙД) і йододефіцитні захворювання (ЙДЗ), що розвиваються на тлі цього стану, за даними ВООЗ, є актуальною проблемою у більш ніж 140 країнах світу, у тому числі, і в Україні. Проблема ЙДЗ визнана актуальною в медико-соціальному аспекті, оскільки ЙД впливає на інтелектуальний потенціал і фізичне здоров’я населення в цілому, крім того, стосується людей будь-якого віку, але найбільше уражає дітей, підлітків та вагітних жінок.
Не випадково, Хартія прав дитини містить, зокрема, право на адекватне йодне забезпечення дитини/майбутньої матері для реалізації генетичного потенціалу, оскільки це необхідна умова активності всіх органів і систем людини протягом усього життя.
Адже йод — есенціальний мікроелемент, основною фізіологічною функцією якого є участь у тиреоїдному синтезі.
Своєю чергою, гормони щитовидної залози регулюють процеси зростання, розвитку, диференціювання, обміну речовин у всіх органах і тканинах організму людини. Дефіцит йоду для дітей та підлітків — це ризик затримки фізичного, психічного та інтелектуального розвитку, розладів статевого дозрівання; порушення функцій ендокринної, серцево-судинної, нервової та імунної систем організму. Для жінок фертильного віку — це неплідність, передчасне переривання вагітності, підвищена материнська та неонатальна смертність, тож говорити про йод і тиреоїдні гормони в рамках конференції, що зібрала педіатрів, потрібно обов'язково, оскільки обмінні процеси найбільш активні, як відомо, в періоди інтенсивного росту, найінтенсивніший з них проходить в утробі матері і триває протягом перших двох років життя. Отже недостатність йоду спричиняє порушення багатьох функцій організму і призводить до розвитку цілого ряду патологічних станів, які у 1983 р. було об’єднано в групу йододефіцитних захворювань (ЙДЗ). Цей термін відображає всі негативні наслідки впливу недостатності йоду на здоров’я людини, але їх можна попередити за умови нормалізації вживання цього мікроелемента. Рекомендовані ВООЗ рівні щоденного споживання йоду наведені у табл. 1.

Отримати цей мікроелемент людина може з продуктів харчування, які містять йод. Максимальна його кількість, як відомо, міститься у морепродуктах, однак, жителі не усіх країн можуть вживати їх щодня, тож для подолання недостатності йоду в харчуванні використовуються методи індивідуальної, групової та масової йодної профілактики. При цьому, масова йодна профілактика є найбільш ефективним і економічним методом усунення дефіциту йоду і досягається шляхом внесення солей йоду (йодиду або йодату калію, йодказеїну) в найбільш вживані продукти харчування: кухонну сіль, хліб, воду, безалкогольні напої, молочні продукти, кондитерські, м'ясні вироби тощо. Отже прийняття національного законодавства щодо йодування солі – найбільш універсальний метод йодної профілактики, здатний без великих матеріально-технічних і фінансових витрат в короткі терміни значно оздоровити населення великих регіонів і практично ліквідувати йододефіцитні захворювання. Швейцарія, Австрія, Італія, Франція та Німеччина першими в світі прийняли таке законодавство (ще більше 100 років тому), адже вже тоді було зрозуміло, що займатися профілактикою значно дешевше і ефективніше, аніж лікуванням. Сьогодні ці країни є взірцем ліквідації проблеми на популяційному рівні.
Щодо України, то за даними моніторування ЙД, що здійснює ВООЗ щодесять років, наша країна знаходиться на п’ятій позиції (знизу!) по рівню вирішення проблеми ЙД. Ми маємо найнижчий рівень вживання йодованої солі в Європі, як результат – кожен десятий український школяр має проблему ЙД. Отже, без прийняття національного законодавства, ми ніколи не подолаємо цю проблему, бо нікого не вмовимо населення пити пігулки, аби поповнити запаси йоду в організмі, протягом всього року, всього життя. Тому адекватної альтернативи йодуванню солі немає, натомість, прийнявши відповідні законодавчі рішення, нам не доведеться говорити про профілактику йодного дефіциту, а отже і про захворювання, ним спричинені.
Доповідь, яка була представлена наступною, була присвячена надзвичайно актуальній темі – проблемам лікування алергічного риніту у дітей. Доповідач – Юрій Гавриленко, к. мед. н., асистент кафедри дитячої оториноларингології, аудіології та фоніатрії НМАПО ім. П. Л. Шупика, докладно зупинився на найважливіших аспектах: сучасні уявлення про алергічний риніт як діагноз, діагностика АР, покази і сучасні стратегії лікування цього захворювання. Зокрема, він нагадав, що маніфестація алергії (і алергічного риніту, зокрема) спостерігається у дуже ранньому віці, ще у період грудного вигодовування, у період, коли в раціон дитини вводяться нові продукти. Саме тоді педіатру нерідко доводиться спостерігати розвиток харчової алергії, яка є предвісником більш загрозливих станів і захворювань, таких, як, наприклад, бронхіальна астма (БА).
Клінічна симптоматика АР добре відома, але, як зауважив лікар, такі симптоми (ринорея, чхання, виділення з носа, утруднення носового дихання) характерні і для інших захворювань верхніх дихальних шляхів, тож на конкретних клінічних випадках лікар продемонстрував, як диференціювати АР з іншими патологіями. Адже вчасно встановлений діагноз і початок лікування – це не тільки профілактика таких ускладнень, як БА, у дитячому віці – це ще й профілактика когнітивних порушень, які можуть виникати на тлі АР. Якісне лікування та профілактики ускладнень РА можливе тільки за умови активної співпраці педіатра, алерголога та отоларинголога, бо тільки отоларинголог здатний виявити всі можливі порушення, які супроводжують патології лор органів, а алерголог може підтвердити або виключити алергічну природу проявів, які так погіршують якість життя маленького пацієнта.
Завершуючи свою доповідь, Юрій Гавриленко нагадав лікарям, що для отримання інформації щодо вирішення проблем навколо цієї патології, варто звертатися до «Національного керівництва з дитячої отоларингології» – видання, яке є гордістю кафедри, яку представляє лікар, і співавтором якого він має честь бути. Він також повідомив, що минулого року вийшла книга, написана співробітниками кафедри під керівництвом професора А. А. Лайко «Алергічні риніти у дітей», де також описані і враховані європейські та американські рекомендації з лікування АР у дітей. Тож, це видання буде вельми корисним для лікарів.
Велика кількість корисної інформації містилася також у доповіді «Вакцинація дітей – проблеми та актуальні питання», яку представив учасникам заходу Федір Лапій, к. мед. н., доцент кафедри дитячих інфекційних хвороб і дитячої імунології НМАПО ім. П. Л. Шупика, гол. дитячий імунолог м. Києва.
Він розпочав свій виступ з констатації невтішного факту: Україна з приводу вакцинації дітей має дуже багато проблем. Наша країна – друга у переліку з кінця (!) серед усіх країн світу за рівнем охоплення вакцинацією дитячого населення. У Сомалі, наприклад, рівень значно вищий, аніж в Україні». На думку експерта, однією з причин такого становища є те, що в нашому суспільстві і досі існує безліч міфів щодо вакцинації. Навіть лікарі та інші медичні працівники нерідко стають «жертвами» таких застарілих уявлень, хоча і знають, що керуватися потрібно лише доказовою медициною, а не міфами, які час від часу з’являються щодо ризиків щеплень, щодо протипоказань та нормальних реакцій, які можуть супроводжувати імунізацію.
Наприклад, частим запитанням до дитячого імунолога є таке: «Чи потрібно перед вакцинацією пройти дегельмінтизацію? І тому подібне. Закон України чітко говорить про те, що вакцинацію мають пройти всі громадяни, що не мають для цього протипоказань: «і не потрібно створювати нових міфів про те, що вакцинувати можна тільки здорових, бо інакше, ми б не говорили про хронічні хвороби. Крім того, важливо розуміти, чому, насправді, існують протипоказання: у тих випадках, коли імунізація не буде ефективною, або становить небезпеку здоров’ю і життю людини (а це дуже рідкісні випадки). Поки ми цього не усвідомимо, так і залишиться, що у нашому Національному протоколі протипокази викладені на 32 сторінках дрібним шрифтом. У США, як і у багатьох країнах Європи, наприклад, ГРІ з легким перебігом не є протипоказанням для вакцинації дитини, а при наявності підвищеної температури вакцинація відкладається до зникнення гострих проявів захворювання. В цьому і є принципова різниця: люди у цивілізованому світі не бояться вакцинації, навпаки, бояться тих наслідків, до яких може призвести її відсутність!».
Упродовж теми імунопрофілактики, головний дитячий імунолог Львівської області, керівник Центру імунології Західноукраїнського спеціалізованого дитячого медичного центру, д. мед. н., проф. каф. клінічної імунології та алергології ЛНМУ ім. Данила Галицького Лариса Костюченко висвітлила у своїй доповіді проблемні питання щодо вакцинації імуноскомпроментованих осіб. Експертна думка з цієї теми дуже важлива для лікарів, тим більше, з уст вченого-клініциста такого високого рівня, адже Центр, яким керує проф. Костюченко, надає високоспеціалізовану медичну допомогу дітям з патологією імунної системи і основними напрямками, в яких спеціалізується Центр, є: первинні імунодефіцити (генетичні дефекти імунної системи, як, наприклад, спадкова агамаглобулінемія, синдром Ніймегена, загальний варіабельний імунодефіцит тощо); обстеження при підозрі на імунодефіцити різного ґенезу та їхній терапевтичний супровід; вакцинація дітей з груп ризику за індивідуальними графіками; алергози (бронхіальна астма, атопічний дерматит, гострі алергічні стани тощо).
Отже, варто сказати, що для більш як 300 учасників заходу з різних куточків України ці 9 годин, протягом яких відбувалася, по суті, інтенсивна робота, принесли чимало користі. Адже педіатри – дуже вдячна аудиторія, спрагла до знань, бо ці спеціалісти звикли серйозно ставитися до удосконалення своїх навичок, розширення ерудиції; звикли використовувати всі можливості для обміну досвідом між колегами. Власне, всім цим вимогам повною мірою відповідала конференція, про яку йшлося.
ДетальнішеСиндром мальабсорбции у детей (часть 2)
В первой части статьи мы рассматривали общие принципы диагностики синдрома мальабсорбции. Далее остановимся на конкретных нозологических формах.
ЦЕЛИАКИЯ (глютен-чувствительная энтеропатия)
За последние десятилетия представления о целиакии претерпели существенные изменения. Раньше считалось, что целиакия – в первую очередь, синдром мальабсорбции, развивающийся у детей раннего возраста вследствие поражения кишечника при употреблении в пищу глютена. Теперь же известно, что целиакия может развиться в любом возрасте и проявляться целым спектром как желудочно-кишечных, так и внекишечных симптомов, а также быть абсолютно бессимптомной. Синдром мальабсорбции – лишь одно из многочисленных проявлений целиакии, причем далеко не самое частое и отмечающееся преимущественно в раннем возрасте.
Согласно современным представлениям, целиакия – хроническое иммуноопосредованное системное заболевание, которое развивается у генетически предрасположенных лиц под действием поступающего с пищей глютена и близких к нему проламинов (белков злаков), характеризующееся вариабельными комбинациями индуцированных глютеномклинических проявлений, а также наличием специфических антител, HLA-DQ2 или DQ8 гаплотипов, и энтеропатии. Специфическими для целиакии антителами являются аутоантитела к тканевой трансглутаминазе 2 (TG2) и эндомизию (EMA), а также антитела к дезаминированным формам пептидов глиадина.
Целиакия – распространенное заболевание (около 1% населения). Наличие генетической предрасположенности к целиакии подтверждается семейной агрегацией и высокой конкордантностью у монозиготных близнецов (почти 100%). Предрасположенность ассоциирована с гаплотипами HLA-DQ2 (DQA1*0501, *0505) и HLA-DQ8 (DQB1*0201, *0202), которые встречаются в популяции с частотой 25% и 30% соответственно. Известно также, что в развитии заболевания могут принимать участие и другие, не HLA-гены, в частности, гены, контролирующие Т-клеточную активацию и рекрутинг (некоторые из них также ответственны за развитие сахарного диабета 1 типа и других аутоиммунных заболеваний).
Целиакия развивается при употреблении в пищу клейковины пшеницы (глютена) и родственных ему проламинов ржи (секалин) и ячменя (гордерин). В большинстве исследований было показано, что употребление овса в плане развития целиакии безопасно, однако, у некоторых больных целиакией обнаруживают активированные проламином овса Т-клетки слизистой оболочки кишечника, которые могут индуцировать воспаление в тонком кишечнике.
Степень риска развития целиакии или возраст начала заболевания у предрасположенных лиц могут изменяться под действием факторов внешней среды. Так, например, позднее введение в рацион глютен-содержащих продуктов, грудное вскармливание, введение небольших количеств глютена способствуют предупреждению развития заболевания у пациентов высокого риска. Кроме того, предполагается роль инфекционных агентов, так как частые ротавирусные инфекции ассоциированы с повышенным риском целиакии.
Целиакия – Т-клеточно-опосредованное хроническое воспалительное заболевание с аутоиммунным компонентом. Выработке аутоантител предшествуют нарушения кишечного пищеварения, изменение проницаемости кишечника и активация механизмов врожденного иммунитета. Иммунодоминантные эпитопы глиадина высокоустойчивы к полостному и пристеночному перевариванию; неполная деградация способствует усилению их иммуностимулирующих и токсических эффектов.
Некоторые пептиды глиадина (p31-43) активируют механизмы неспецифического иммунитета, в частности, индуцируют выработку интерлейкина (ИЛ) 15. Последний, так же, как и интерфероны типа 1, может изменять толерогенный фенотип дендритных клеток, что приводит к активации Т-клеток в собственной пластине пептидами, связанными с HLA-DQ2 или HLA-DQ8 молекулами. Активность глиадин-специфических Т-клеток усиливается благодаря действиюTG2; этот фермент дезаминирует пептиды глиадина, что приводит к повышению их аффинности для HLA-DQ2 или HLA-DQ8. Основными цитокинами, которые вырабатываются вследствие активации клетокглиадином, являются интерфероны (сдвиг в сторону образования T-хелперов 1 типа), а также ИЛ 21. Результатом воздействия активированных Т-лимфоцитов становится ремоделирование слизистой оболочки с участием металлопротеиназ и факторов роста, что приводит к формированию классической «плоской слизистой оболочки».Также при целиакии отмечаются значительные нарушения в гомеостазе интраэпителиальных лимфоцитов. ИЛ 15 усиливает экспрессию рецепторов естественных киллеров CD94 и NKG2D, а также эпителиальных стрессовых молекул, что способствует усилению цитотоксичности, активации апоптоза клеток и развитию атрофии ворсинок.
Наиболее очевидным проявлением аутоиммунитета при целиакии является наличие в сыворотке крови антител к TG2. Однако механизмы аутоиммунной реакции по большей мере неизвестны, как и их патогенетическое значение. Так, возможность «потенциальной» целиакии, когда антитела к TG2 обнаруживаются, а слизистая кишечника не изменена, указывает на то, что образование антител не обязательно сопровождается повреждением кишечника.
Клинические проявления
Проявления целиакии разнообразны (табл. 1). Наличие манифестной кишечной симптоматики наиболее характерно для детей, у которых заболевание диагностировано в течение первых 2 лет жизни. В большинстве таких случаев отмечают задержку физ. развития, хроническую диарею, рвоту, вздутие живота, атрофию мышц, анорексию и раздражительность. Иногда могут возникать запор, выпадение прямой кишки или инвагинация. С внедрением серологических скрининговых тестов у пациентов разных возрастных групп стало очевидно, что нарушения при целиакии могут затрагивать практически все органы. Основные внекишечные проявления целиакии:
- Железодефицитная анемия, резистентная к пероральной ферротерапии – наиболее частое внекишечное проявление. В детском возрасте железодефицит редко бывает единственным проявлением заболевания.
- Низкий рост и задержка полового созревания. У приблизительно 10% детей с идиопатической низкорослостью может быть диагностированацелиакия. При этом обнаруживаемая у части этих пациентов сниженная стимулированная продукция гормона роста нормализуется после перехода на безглютеновую диету.
- Хронический гепатит и гипертрансаминаземия. У пациентов с нелеченной целиакией часто отмечается повышение уровней АлАТ и АсАТ. Среди пациентов с гипертрансаминаземиями невыясненной этиологии около 9% могут иметь целиакию. При биопсии печени у этих пациентов определяются признаки неспецифического реактивного гепатита. На безглютеновой диете в большинстве случаев уровень ферментов печени в крови нормализуется.
- Артрит и артралгии. Среди детей с ювенильным хрон. артритом не менее 3% могут иметь целиакию.
- Oстеопения и остеопoроз. К моменту установления диагноза у 50% детей и 75% взрослых с целиакией отмечается снижение мин. плотности костей, иногда вплоть до остеопороза. На фоне диеты без глютена минерализация постепенно восстанавливается: у детей – примерно через год.
- Герпетиформный дерматит – буллезная сыпь, локализующаяся в области локтей, коленей и ягодиц, ассоциированная с кожными гранулярными IgА-депозитами. Высыпания уменьшаются и проходят на фоне безглютеновой диеты.
- Гипоплазия зубной эмали постоянных зубов.
- Неврологические проблемы более характерны для взрослых. Целиакия может вызвать тяжелую эпилепсию с затылочными кальцификатами. Такие пациенты обычно плохо отвечают на противосудорожные препараты, а при назначении безглютеновой диеты отмечается улучшение (если она начата вскоре после первых приступов). Также хорошо описана связь между целиакией и мозжечковой атаксией у взрослых.
- Психиатрические расстройства. Хотя возможная связь с целиакией рассматривалась при множестве поведенческих расстройств (например, при аутизме, синдроме гиперактивности с дефицитом внимания), доказательств ее наличия получено не было. Однако при целиакии могут отмечаться депрессия и тревожность. Эти состояния могут быть серьезными и обычно уменьшаются на фоне безглютеновой диеты.
- Другие внекишечные проявления включают периферические невропатии, кардиомиопатию, афтозный стоматит и алопецию. Механизмы, определяющие характер и тяжесть клин. проявлений, неясны. Возможными причинами считают нарушения всасывания различных нутриентов и аномальные иммунные реакции.
Заболевания, ассоциированные с целиакией

Некоторые заболевания, в основном – аутоиммунные, обнаруживаются у пациентов с целиакией чаще, чем в популяции. Среди них сахарный диабет 1 типа, аутоиммунные заболевания щитовидной железы, болезнь Аддисона, синдром Шегрена, аутоиммунный холангит, аутоиммунный гепатит, первичный билиарный цирроз. Такие ассоциации, по всей видимости, обусловлены идентичными HLA-гаплотипами. Другие связанные с целиакией состояния включают селективный дефицит IgA, синдромы Дауна, Шерешевского–Тернера и Вильямса.
В зависимости от характера клинических проявлений, наличия энтеропатии и серологических маркеров выделяют несколько клинических форм целиакии:
- типичная – наличие выраженных гастроинтестинальных симптомов, характерных морфологических изменений в тонкой кишке (энтеропатии), специфических для целиакии антител и HLA-гаплотипов;
- атипичная – гастроинтестинальные симптомы выражены слабо или отсутствуют, а в клинике доминирует внекишечная симптоматика; при этом выявляются специфические антитела, HLA-гаплотипы и энтеропатия;
- скрытая («тихая») – клинические симптомы отсутствуют, но обнаруживаются энтеропатия, специфические антитела и HLA-гаплотипы;
- латентная – гистологических и серологических маркеров нет, но у пациента была диагностирована целиакия когда-либо ранее; клинические симптомы могут присутствовать или нет;
- потенциальная – морфология слизистой тонкого кишечника не изменена, выявляются специфические антитела и HLA-гаплотипы; симптомы могут присутствовать или нет; в будущем энтеропатия может развиться или нет.
Своевременная диагностика бессимптомных форм целиакии, частота которых значительно превышает частоту манифестных форм, важна, так как у пациентов с целиакией, даже бессимптомной, отмечается повышенная смертность, причем риск смертности выше при поздней диагностике заболевания и/или несоблюдении диеты. Основная причина смерти – неходжкинская лимфома. У взрослых могут развиваться такие осложнения как рефрактерная целиакия, язвенный еюноилеит и обусловленная энтеропатией Т-клеточная лимфома.
Диагностика
Диагностика целиакии базируется на оценке совокупности клинических симптомов, данных серологических тестов, HLA-типирования и гистологического исследования дуоденальных биоптатов. В январе 2012 года были опубликованы рекомендации ESPGHAN (Европейское Общество педиатрической гастроэнтерологии, гепатологии и питания) по диагностике целиакии у детей. Отличием данного руководства от предшествовавших американских (NASPGHAN, 2004) и британских (NICE, 2008) рекомендаций стало выделение условий, при которых проведение биопсии тонкой кишки для диагностики целиакии не обязательно.
Наиболее чувствительными и специфичными серологическими тестами для диагностики целиакии являются определение уровней антител к TG2 и эндомизию, а также антител к дезаминированному пептиду глиадина. У пациентов с нормальным уровнем IgA предпочтение отдается определению указанных антител, относящихся к классу IgA. При снижении уровня общего IgA в сыворотке крови (менее 0,2 г/л) определяют специфические антитела класса IgG, хотя они и менее специфичны. Важным условием является определение уровней специфических антител ДО назначения безглютеновой диеты, т. к. на фоне ее титры антител снижаются. Определение уровня антиглиадиновых антител (антител к глиадину) в крови в настоящее время не рекомендуется в связи с недостаточной чувствительностью и специфичностью метода.
Гистологическое исследование биоптатов тонкой кишки. Приоритет этого метода в диагностике целиакии был подвергнут сомнению, так как оказалось, что выявляемые изменения слизистой оболочки не являются специфичными для целиакии, а могут наблюдаться и при некоторых других заболеваниях (табл. 2). Кроме того, эти изменения неоднородны, могут локализоваться только в луковице двенадцатиперстной кишки, а выявление их зависит как от соблюдения процедуры подготовки образцов, так и от опыта исследователя.
Биопсия проводится во время ФЭГДС; по крайней мере, 1 образец должен быть взят из луковицы дуоденум и не менее 4 – из ниже лежащих отделов двенадцатиперстной кишки.
Степень изменений слизистой оболочки тонкой кишки определяется в соответствии с классификацией Marsh-Oberhuber:
- Marsh 0 – преинфильтративная стадия (норма)
- Marsh I – инфильтративные изменения – структура слизистой оболочки нормальная, повышено количество интраэпителиальных лимфоцитов (более 25 на 100 энтероцитов);
- Marsh II – гиперпластические изменения – то же + гиперплазия крипт; снижение соотношения «высота ворсин/глубина крипт» (в норме 3-5 : 1);
- Marsh III – деструктивные изменения – изменения Marsh II + атрофия ворсин:
- Marsh III A – парциальная атрофия ворсин: соотношение «высота ворсин / глубина крипт» меньше 1;
- Marsh III B – субтотальная атрофия ворсин: отдельные ворсины еще распознаваемы;
- Marsh III C – тотальная атрофия ворсин: слизистая без пальцевидных возвышений, напоминает слизистую оболочку толстой кишки.
HLA-типирование. Отсутствие HLA-DQ2- и HLA-DQ8-гаплотипов позволяет полностью исключить целиакию, равно как и возможность ее развития в будущем. HLA-типирование может быть полезным в сомнительных случаях – например, если гистологически определяется умеренная энтеропатия (Marsh I–II), а специфические антитела не выявляются.
Несмотря на широкую распространенность целиакии и доступность скрининговых тестов, массовый скрининг не рекомендуется. Обследованию на целиакию подлежат:
1) пациенты с симптомами, подозрительными в отношении целиакии:
- иначе не объясненными хронической или рецидивирующей диареей,
- задержкой физического развития,
- потерей массы тела,
- низкорослостью,
- задержкой полового созревания,
- аменореей,
- железодефицитной анемией,
- тошнотой и рвотой,
- хроническими болями в животе,
- вздутием живота,
- запорами,
- хронической усталостью,
- рецидивирующим афтозным стоматитом,
- герпетиформным дерматитом,
- отклонениями в печеночных биохимических пробах,
- переломами костей, неадекватными травмам/остеопенией/остеопорозом.
2) бессимптомные пациенты с повышенным риском развития целиакии (с ассоциированными заболеваниями):
- сахарным диабетом типа 1,
- синдромом Дауна,
- аутоиммунными заболеваниями щитовидной железы,
- синдромом Шерешевского–Тернера,
- синдромом Вильямса,
- селективным дефицитом IgA,
- аутоиммунным гепатитом,
- родственники больных целиакией первой степени родства.
У симптоматических пациентов обследование начинают с определения уровня антител к тканевой трансглутаминазе 2 (анти-TG2) IgA в сыворотке крови, а также общего IgA для исключения его дефицита. Альтернативой определению общего IgA может быть тестирование на уровень антител к дезаминированному пептиду глиадина (анти-DGP) класса IgG. Если получен отрицательный результат анти-TG2 IgA при нормальном уровне общего IgA (или отрицательном результате анти-DGPIgG), диагноз целиакии маловероятен, кроме нескольких особых ситуаций (возраст младше 2 лет, соблюдение безглютеновой диеты на момент обследования, тяжелое течение заболевания, наследственная предрасположенность или наличие заболеваний, предрасполагающих к целиакии, иммуносупрессивная терапия). У детей младше 2 лет анти-DGP могут быть более чувствительным маркером, чем анти-TG2.
Если результат исследования на анти-TG2 положительный, то пациент должен быть направлен к гастроэнтерологу для проведения дальнейшего обследования, объем которого зависит от уровня анти-TG2. При уровне антител к TG2 менее 10 значений верхней границы нормы проводится эндоскопическое исследование с биопсией тонкой кишки. Если уровень анти-TG2 превышает 10 значений верхней границы нормы, назначают тестирование на анти-ЕМА и HLA-типирование. В случае выявления DQ2 или DQ8, а также положительного теста на анти-ЕМА у таких пациентов диагноз целиакии можно считать подтвержденным без проведения биопсии. Если же у пациентов с высокими титрами анти-TG2 (более 10 норм) не выявляют характерных HLA-гаплотипов и/или результат анти-ЕМА оказывается отрицательным, рассматривают возможность ложно-положительных и ложно-отрицательных результатов. Для этого обычно проводят повторные серологические тесты и дуоденальную биопсию.
Во всех руководствах по диагностике целиакии подчеркивается, что безглютеновая диета не должна назначаться до завершения диагностического процесса и получения убедительных данных в пользу целиакии.
В серонегативных случаях у пациентов с тяжелым течением заболевания и характерными для целиакии симптомами необходимо проведение тонкокишечной биопсии и HLA-типирования.
Возможна ситуация, когда в ходе обследования по поводу каких-либо гастроентерологических симптомов была проведена дуоденальная биопсия, и при морфологическом исследовании биоптатов обнаружены характерные для целиакии изменения слизистой оболочки тонкой кишки (Marsh I–III). В таких случаях для подтверждения диагноза следует определить уровень анти-TG2, а для детей младше 2 лет – анти-DGP и провести HLA-типирование.
У бессимптомных пациентов с повышенным риском развития целиакии в качестве первичного скрининга рекомендуется проведение HLA-типирования. При отсутствии у ребенка HLA-DQ2 и HLA-DQ8 не имеет смысла обследовать его на целиакию ни теперь, ни когда-либо в будущем.
В случае выявления HLA-DQ2 или HLA-DQ8-гаплотипа рекомендуется периодически определять титры специфических для целиакии антител (анти-TG2) в крови (но не ранее, чем ребенку исполнится 2 года).
Поскольку известно, что у лиц с повышенным генетическим риском развития целиакии титры антител могут колебаться, а классических клинических симптомов заболевания может и не быть, без гистологического подтверждения диагноз целиакии невозможен.
Если при первичном серологическом обследовании бессимптомных пациентов уровень специфических антител (анти-TG2 или анти-DGP) повышен незначительно (менее 3 норм), следует определить уровень анти-ЕМА в крови. В случае позитивного теста анти-ЕМА показана дуоденальная биопсия. Если тест на анти-ЕМА отрицателен, рекомендуется повторное серологическое тестирование через 3–6 месяцев при условии, что рацион пациента будет содержать достаточное, адекватное возрасту, количество глютена.
Лечение
Лечение целиакии состоит в соблюдении пожизненной безглютеновой диеты. Из рациона должны быть исключены продукты, содержащие пшеницу, ячмень и рожь. В последнее время было доказано, что употребление овса безопасно для пациентов с целиакией, однако существуют опасения по поводу контаминации овса глютеном во время сбора, помола и транспортировки. Тем не менее, если овсяные продукты прошли соответствующую проверку и признаны свободными от глютена, они должны использоваться.
Придерживаться безглютеновой диеты должны все пациенты с установленным диагнозом, независимо от наличия симптомов. У пациентов с симптоматической целиакией на фоне диеты без глютена клиническое улучшение обычно наступает в течение нескольких недель. У бессимптомных пациентов оценить эффективность безглютеновой диеты сложнее. В таких случаях можно ориентироваться по отсутствию признаков дефицита нутриентов, и в первую очередь остеопении, при длительном наблюдении. Нормализация уровня антител должна произойти в течение 12 месяцев с момента перехода на безглютеновую диету. В настоящее время недостаточно данных о рисках для здоровья у нелеченных пациентов с незначительной энтеропатией, которые могут быть клинически бессимптомными. Также нет указаний относительно необходимости безглютеновой диеты у пациентов с «потенциальной»целиакией.
Безглютеновыми считаются продукты, содержащие менее 20 ppm глютена. Количество глютена, которое могут безопасно употреблять больные целиакией, пока окончательно не определено, но считается, что пороговым значением должно быть менее 50 мг/сут,. хотя индивидуальная изменчивость затрудняет установление универсального порога. Важно, чтобы опытный диетолог, обладающий специальными знаниями в консультировании по целиакии, обучал семью и ребенка ограничениям в питании.
Соблюдение диеты без глютена может быть трудной задачей, особенно для подростков. В качестве контроля рекомендуется периодически оценивать наличие симптомов, темпы роста и физического развития. Измерение уровней анти-TG2 в крови может быть полезно как маркер комплаенса, хотя при легких нарушениях диеты они не показательны.

Гиперчувствительность к глютену без целиакии
Помимо целиакии, в настоящее время выделяют еще 2 расстройства, связанных с употреблением в пищу глютена.
IgE-опосредованная аллергия на пшеницу может иметь сходные с целиакией симптомы, однако чаще встречается без энтеропатии, с симптомами атопии (крапивница, ангиоедема, экзема, астма, ринит) и диагностируется по наличию специфических IgE к пшенице (кожный прик-тест или уровень специфических IgE в крови). Отличием данного состояния является также быстрое развитие симптомов, т. е. в течение нескольких минут или часов после употребления пшеницы.
О непереносимости глютена без целиакии говорят при наличии различных комбинаций кишечных (боль в животе, вздутие живота, диарея) и внекишечных (мигрень, хроническая усталость, мышечные боли, высыпания) симптомов, связанных с приемом глютен-содержащих продуктов, но при отсутствии энтеропатии и характерных для целиакииа утоантител и HLA-гаплотипов. Симптомы часто напоминают клинику синдрома раздраженного кишечника, и у некоторых пациентов с синдромом раздраженного кишечника состояние улучшается на фоне безглютеновой диеты. Непереносимость глютена без целиакии в детском возрасте изучена недостаточно, большая часть исследований касалась взрослых. В любом случае непереносимость глютена без целиакии – диагноз исключения, когда нет данных, подтверждающих наличие целиакии и аллергии на пшеницу, но отмечается отчетливое улучшение на фоне диеты без глютена. Кроме того, пока непонятно, насколько строгой должна быть диета у таких пациентов, какое количество глютена они способны безопасно переносить, как долго нужно соблюдать диету, как оценивать ее эффективность, нужно ли ограничивать употребление продуктов из ржи и ячменя, и, наконец, является ли это заболевание пожизненным, как целиакия, или же транзиторным, со спонтанным выздоровлением.
Продолжение в следующем номере
Детальніше