

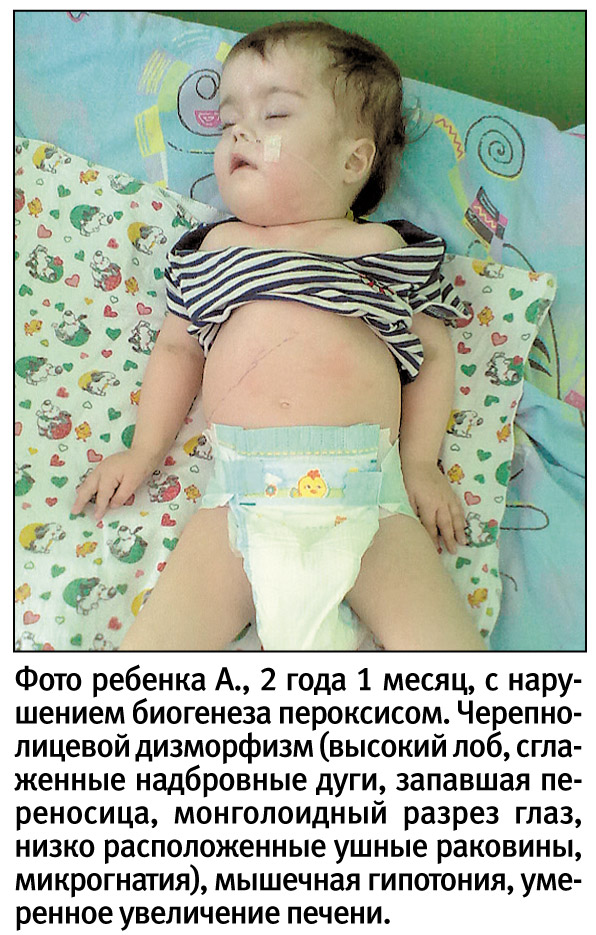
 Ребенок А., девочка, родилась от IV беременности, осложненной угрозой прерывания в первой половине, вульвовагинитом на 14-й неделе. Роды II в сроке 40 недель, со стимуляцией. Оценка по шкале Апгар 7/8 баллов, масса тела при рождении 2750 г, длина тела — 51 см, окружность головы 32 см, окружность груди 31 см. Родители и старший ребенок здоровы.
Ребенок А., девочка, родилась от IV беременности, осложненной угрозой прерывания в первой половине, вульвовагинитом на 14-й неделе. Роды II в сроке 40 недель, со стимуляцией. Оценка по шкале Апгар 7/8 баллов, масса тела при рождении 2750 г, длина тела — 51 см, окружность головы 32 см, окружность груди 31 см. Родители и старший ребенок здоровы.
С первых дней жизни отмечалась мышечная слабость, заторможенность, что обуславливало затруднения при вскармливании — самостоятельно сосать грудь девочка не могла (кормилась сцеженным молоком из бутылочки). Также имели место: увеличение размеров большого родничка до 4x4 см, расхождение сагиттального шва до 1 см, открытый малый родничок 1x1 см. Указанные изменения расценивались как последствия перинатального поражения ЦНС. Из родильного дома ребенок был выписан на 10-е сутки.
В возрасте 2 недель жизни появились геморрагические расстройства в виде подкожных гематом на пояснице и грудной клетке и периодических кровянистых выделений из носа, а также умеренно выраженный отечный синдром. Проведенное обследование выявило наличие гипопротеинемии (уровень общего белка — 20 г/л), увеличение времени свертываемости крови (более 15 минут) при нормальном уровне тромбоцитов, снижение ПТИ до 55%, а также синдром цитолиза (АЛТ — 7 норм, АСТ — 6 норм) в сочетании с нормальным уровнем билирубинемии, умеренным увеличением печени и нормальными размерами селезенки. Данное состояние было расценено как проявления ВУИ, фетальный гепатит. Однако результаты проведенного тестирования на инфекции TORCH-группы, маркеры вирусных гепатитов были отрицательными. Параметры коагуляционного гемостаза и уровень белка в крови быстро нормализовались после переливания свежезамороженной плазмы, а явления цитолиза оставались резистентными к проводимой терапии.
В дальнейшем, в течение 2–4-го месяцев жизни в клинике доминировала грубая задержка психомоторного и физического развития. Ребенок не приобретал навыков, не реагировал на звук и свет, сохранялось увеличение размеров родничков и расхождение сагиттального шва, мышечная гипотония, отмечался нистагм, имела место пропорциональная задержка роста и прибавки массы тела. В возрасте 4 месяцев была диагностирована частичная атрофия зрительных нервов, двусторонняя нейросенсорная тугоухость. В течение всего этого времени сохранялось 5–6-кратное повышение уровня трансаминаз в сыворотке крови.
В возрасте 5 месяцев ребенок был обследован в клинике «ОХМАДЕТ». Выявлены синдром цитолиза, повышение в крови уровней лактата, КФК, 5-оксипролина, в моче периодически обнаруживались аминокислоты: аланин, глицин, лейцин. По данным МРТ — кистозно-глиозные изменения на уровне подкорковых структур слева, признаки двухстороннего отита. Был установлен диагноз: «Криптогенный гепатит с минимальной активностью, без нарушения функции печени. Грубая задержка психомоторного развития. Спастический тетрапарез. Врожденный нистагм. Частичная атрофия зрительных нервов обоих глаз. Тугоухость».
В то же время было отмечено наличие у ребенка множественных стигм дизэмбриогенеза (башенный череп, высокий лоб, уплощенный профиль лица, монголоидный разрез глаз, уплощение спинки носа, микрогнатия, узкие носовые ходы и слуховые проходы), что в совокупности с признаками дегенеративного поражения мозга и вовлечения в патологический процесс паренхиматозных органов (печень, почки?) наводило на мысль о возможном метаболическом генезе выявленных изменений.
В возрасте 1 года 7 месяцев девочку обследовали в Медико-генетическом научном центре (Москва), в лаборатории наследственных болезней обмена веществ. Было выявлено повышение концентраций в крови жирных кислот с очень длинной цепью (С26), повышение соотношения С24/С22 и С26/С22 жирных кислот, нормальные уровни бета-D-галактозидазы, гексозаминидазы А. Методом прямого секвенирования проведено частичное исследование кодирующих экзонов гена РЕХ1. В экзоне 15 выявлена патогенная мутация с.2528G>А, приводящая к замене аминокислоты p.Gly843Asp в гетерозиготном состоянии. На основании клинических и лабораторных данных выставлен диагноз: «Синдром Цельвегера».
Синдром Цельвегера – одна из клинических форм группы наследственных заболеваний, в основе которых лежит нарушение биогенеза пероксисом (НБП).
Пероксисомы – клеточные органеллы, присутствующие во всех эукариотических клетках (кроме эритроцитов), функциональная активность которых преимущественно связана с окислением жирных кислот с очень длинной цепью (более 22 атомов углерода). Пероксисомы принимают участие в синтезе ряда важных веществ: желчных кислот, докозагексаеновой кислоты, плазмалогенов. Кроме того, в пероксисомах утилизируется кислород с образованием перекиси кислорода, которая используется затем при окислении токсичных веществ.
В биогенезе пероксисом участвуют специальные белки – пероксины. Сегодня известно не менее 32 пероксинов (из них у человека около 14), которые кодируются группой генов РЕХ. Дефекты этих генов и лежат в основе пероксисомных болезней.
Поражение нервной системы, органов зрения и слуха, задержка физического развития при НБП обусловлены, с одной стороны, дефицитом докозогексаеновой кислоты, а с другой – накоплением жирных кислот с очень длинной цепью (ОДЦЖК) и фитановой кислоты. Уменьшение количества рецепторов к АКТГ в надпочечниках из-за изменений физико-химических свойств мембран, связанных с накоплением ОДЦЖК, может привести к надпочечниковой недостаточности. Геморрагические расстройства в периоде новорожденности при НБП обусловлены дефицитом витамин-К-зависимых факторов свертывания, нарушением печеночных функций, дефицитом плазмалогенов.
Описано 4 клинических варианта НБП: синдром Цельвегера, неонатальная адреналолейкодистрофия, инфантильная болезнь Рефсума и ризомелическая точечная хондродисплазия. При этом первые три варианта – разные степени тяжести одного и того же заболевания. Частота НБП в популяции – 1:50000–100000 живорожденных. Тип наследования – аутосомно-рецессивный.
Синдром Цельвегера впервые был описан в 1964 году и включает:
- комплекс врожденных черепно-лицевых дизморфий – уплощенное лицо и затылок, высокий лоб, гипертелоризм, эпикант, мелкие орбиты, гипоплазия надбровных дуг, «готическое» небо, широкая запавшая переносица, микрогнатия, низко расположенные уши, избыточные складки на шее, расхождение черепных швов;
- поражение нервной системы – мышечная гипотония, гипорефлексия, грубая задержка ПМР, судороги, на КТ и ЯМР ГМ – множественные мелкие перивентрикулярные кисты, диффузная демиелинизация вдоль сильвиевой борозды и фронтопариетальной области, пахигирия, полимикрогирия;
- поражение глаз – нистагм, атрофия зрительных нервов, катаракта, глаукома, пигментная ретинопатия;
- нарушение слуха – нейросенсорная тугоухость;
- поражение внутренних органов – гепатомегалия, желтуха, мальабсорбция жиров, поликистоз почек, отставание в физическом развитии, истощение;
- поражение костей – зернистая кальцификация надколенников и эпифизов длинных костей, синхондроз вертлужных впадин, остеопороз.
Симптомы при синдроме Цельвегера выражены уже с рождения, дети редко доживают до 1 года.
 При неонатальной адреналолейкодистрофии менее выражены черепно-лицевые дизморфии, отсутствуют поликистоз почек и зернистая кальцификация надколенников. Заболевание манифестирует во втором полугодии жизни. Основные проявления – поражение нервной системы, органов зрения и слуха, печени. Продолжительность жизни – около 3 лет, иногда до 10 лет.
При неонатальной адреналолейкодистрофии менее выражены черепно-лицевые дизморфии, отсутствуют поликистоз почек и зернистая кальцификация надколенников. Заболевание манифестирует во втором полугодии жизни. Основные проявления – поражение нервной системы, органов зрения и слуха, печени. Продолжительность жизни – около 3 лет, иногда до 10 лет.
Инфантильная болезнь Рефсума манифестирует после 1 года. Черепно-лицевые дизморфии отсутствуют. В клинике доминирует поражение нервной системы (нарушения интеллектуального и моторного развития разной степени тяжести), слуха и зрения. Некоторые больные доживают до 2-го десятилетия.
В диагностике НБП ключевым моментом является обнаружение высокого уровня ОДЦЖК, фитиновой и пипеколиновой кислот в крови. Для дифдиагностики НБП и изолированных пероксисомных дефектов определяют уровень ацилкарнитинов с очень длинной цепью в крови.
При молекулярно-генетическом анализе чаще всего обнаруживают мутации в гене РЕХ1: 25–37% – с.2528G>А (15 экзон), около 30% – с.2097_2098insT (13 экзон). Эти 2 мутации отвечают за приблизительно 40% всех случаев НБП.
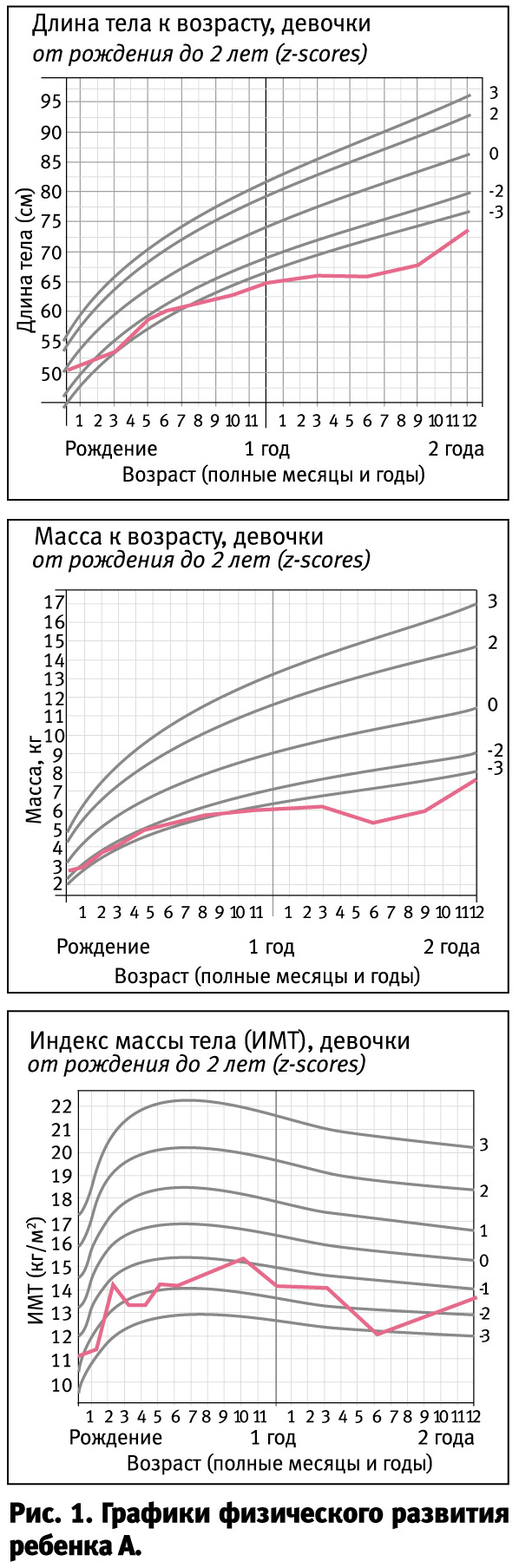
В настоящее время девочке 2 года 1 месяц. В клинике доминируют неврологические проявления – грубая задержка ПМР, мышечная гипотония и судороги по типу инфантильных спазмов, появившиеся после 1,5 лет и плохо поддающиеся терапии антиконвульсантами, а также поражение органов слуха, зрения и печени. К 2 годам уровень трансаминаз нормализовался (цирроз печени?). Масса тела 7450 г, длина – 74 см.
Таким образом, у ребенка имеет место нарушение биогенеза пероксисом, что было подтверждено высоким уровнем ОДЦЖК и обнаружением специфической мутации в гене РЕХ1. Клиническая форма скорее соответствует неонатальной адреналолейкодистрофии (отсутствие поликистоза почек и костных изменений, нерезко выраженный дизморфизм), хотя имело место раннее начало симптомов. Редкость заболевания и ограничение диагностических возможностей обусловили относительно позднюю диагностику в данном случае.
Литература
- Зиновник А.Б., Гусина Н.Б. Нарушение биогенеза пероксисом (клиника, диагностика, лечение). Методические рекомендации для врачей. – Минск, 2011. – 30 с.
- Берман Р.Э. Педиатрия по Нельсону: в 5 т.: пер. с англ. – Т.2. – М.: ООО «Рид Элсивер», 2009. – С. 402-410.
- Abdel-Hamid HZ. Peroxisomal Disorders. – Режим доступа: http://emedicine.medscape.com/article/1177387-overview
коментарів