ГІПЕРПАРАТИРЕОЗ
Класифікація МКХ 10: Е 21.
Визначення
Гіперпаратиреоз – гетерогенна група станів, що характеризується гіперфункцією прищитоподібних залоз (ПЩЗ).
Патогенетично виділяють: первинний, вторинний і третинний гіперпаратиреоз.
В основі первинного гіперпаратиреозу лежить гіперплазія або аденома ПЩЗ. Він характеризується поєднанням високої концентрації паратгормону (ПТГ) з гіперкальціємією. Вторинний гіперпаратиреоз – нормальна реакція ПЩЗ на тривалу гіпокальціємію у зв'язку дефіцитом вітаміну D або із захворюваннями, що вражають нирки, печінку, кишківник. Вторинний гіперпаратиреоз проявляється високою концентрацією ПТГ і гіпокальціємією. Третинний гіперпаратиреоз виникає у пацієнтів з давнім вторинним гіперпаратиреозом і супроводжується підвищеною концентрацією ПТГ в сироватці крові та появою гіперкальціємії.
Первинний гіперпаратиреоз (Е 20.0)
Це синдром, що обумовлений надлишковою продукцією ПТГ за рахунок ураження безпосередньо прищитоподібних залоз, наприклад гіперплазії або аденоми, та супроводжується гиперкальціємією.
Це третя за розповсюдженістю ендокринологічна патологія, частота якої варіює від 0,5 до 34 на 1000 населення у залежності від методів виявлення.
Первинний гіперпаратиреоз в середньому діагностується з частотою 25 нових випадків на 100 тис. населення на рік. З ним пов'язано 20% випадків синдрому гіперкальціємії.
Переважна більшість аденом ПЩЗ (80% випадків), з якими і пов'язують розвиток первинного гіперпаратиреозу, розташовуються у типовому місці – за щитоподібною залозою.
У дітей гіперпаратиреоз зустрічається рідко і виникає, як правило, внаслідок розвитку доброякісної аденоми.
Зазвичай клінічні прояви з'являються після 10-річного віку.
Етіологія
Основні причини первинного гіперпаратиреозу:
1) солітарна (80%) або множинні (5%) аденоми ПЩЗ;
2) гіперплазія ПЩЗ (15%);
3) карцинома ПЩЗ (<5%);
4) первинний гіперпаратиреоз у межах синдромів множинних ендокринних неоплазій 1-го та 2-го типу (МЕН-1, МЕН-2).
Патогенез
Підвищена продукція паратгормону призводить до надмірного виведення нирками фосфатів. Зниження сироваткового рівня останніх стимулює синтез 1,25-(OH)2-D3, який сприяє всмоктуванню надлишку Ca2+ у кишківнику. Далі гіперкальціємія підсилюється за рахунок активації остеокластів надлишком паратгормону. Збільшена кількість паратгормону призводить до прискорення обміну в кістках, прискорення кісткової резорбції та кісткового утворення. Але формування нової кісткової тканини відстає від її розсмоктування, що викликає генералізований остеопороз та остеодистрофію, вимивання кальцію з кісткових депо та гіперкальцемію. Також виникає кальціурія, наслідком якої є пошкодження епітелію ниркових канальців, а також утворення каменів у нирках. Нефрокальціноз, в свою чергу, веде до порушення функцій нирок. Підвищення рівнів концентрації кальцію і фосфатів у сироватці крові можуть привести до кальцифікації судин кровоносного русла, обумовлюючи ризик передчасної смерті.
Клініка
Основними скаргами хворих є стомлюваність, м'язова слабкість, біль у кістках, зниження апетиту, нудота, блювота. Більш ніж в 50% випадків діагноз встановлюється при випадковому виявленні гіперкальціємії. Клінічні прояви хвороби пов'язані з рівнем гіперкальціємії. При рівні концентрації кальцію у сироватці крові 3,0–3,5 ммоль/л виникають нездужання і сонливість, 3,5–4 ммоль/л — можливий розвиток психозу, а при концентрації кальцію в сироватці крові понад 4 ммоль/л спостерігається порушення свідомості, розвиток коми і можливий летальний результат.
Симптоматика первинного гіперпаратиреозу складається з наступних груп симптомів:
1) Ниркова симптоматика (40–50%). Нефролітіаз, рідше – прогностично несприятливий нефрокальціноз. Рефрактерний до дії антидіуретичного гормону інсипідарний синдром (поліурія, полідипсія, гіпоізостенурія), який у важких випадках призводить до ниркової недостатності.
2) Кісткова симптоматика (50%). Гіперпродукція паратгормону призводить до негативного кісткового балансу. Рентгенологічно виявляється дифузна остеопенія: при дослідженні кистей – у 40% випадків, хребців – у 20%. У важких випадках може встановлюватися патогномонічна субперіостальна резорбція і акроостеоліз кінцевих фаланг кистей і стоп. Кісти, гігантськоклітинні пухлини і епуліди у даний час виявляються винятково рідко.
3) Гастроінтестинальна симптоматика (50%). Анорексія, нудота, обстипація, метеоризм, схуднення, у 10% випадків мають місце пептичні виразки шлунку та/або дванадцятипалої кишки, у 10% – панкреатит (при загостренні рівень Са2+ може знижуватися), рідше панкреакалькульоз. Удвічі частіше, ніж у популяції, зустрічається жовчнокам'яна хвороба.
4) Нейром'язова симптоматика. М'язова слабкість і атрофія, скорочення інтервалу QT на ЕКГ. Депресія, сонливість, ослаблення пам'яті.
5) Ураження серцево-судинної системи при гіперпаратиреозі характеризується артеріальною гіпертензією, гіпертрофією лівого шлуночка, кальцифікацією коронарних артерій та клапанів серця.
6) Гіперкальціємічний криз – важке, загрозливе для життя ускладнення, що зустрічається менш ніж у 5% пацієнтів. Провокується гіподинамією, переломами кісток, призначенням тіазидних діуретиків. Клінічно гіперкальціємічний криз проявляється поліурією, полідипсією, блювотою, ексикозом, адинамією, сомноленцією, комою.
Первинний гіперпаратиреоз у новонароджених
Тяжкий первинний гіперпаратиреоз новонароджених – рідкісний розлад. В більшості пацієнтів є мутації гена CASR. Симптоми розвиваються незабаром після народження. Основними проявами є анорексія, дратівливість, млявість, закріпи, недостатня прибавка маси тіла. У новонароджених із важким гіперпаратиреозом спостерігається помітне підвищення кальцію і ПТГ у сироватці крові. Концентрація ПТГ дуже висока, а гіперкальціємія небезпечна для життя. Рентгенологічно визначається підокістна резорбція кістки, остеопороз, патологічні переломи. Гістологічно визначається дифузна гіперплазія прищитоподібних залоз. Зазвичай первинний гіперпаратиреоз новонароджених є спадковим ураженням. Діти з важким гіперпаратиреозом новонароджених можуть бути гомозиготними або гетерозиготними за мутацією гена кальцієвих каналів, у той самий час більшість людей з цією мутацією мають аутосомно-домінантну сімейну гіперкальціємію.
Множинні ендокринні неоплазії
МЕН I типу є аутосомно-домінантним захворюванням, яке характеризується гіперплазією або неоплазією ендокринної частини підшлункової залози (що виділяє гастрин, інсулін, панкреатичний поліпептид, а іноді і глюкагон), передньої долі гіпофіза (що зазвичай виділяє пролактин) і прищитоподібних залоз. У більшості випадків МЕН I зустрічається після 50-річного віку, і лише в рідкісних випадках у дітей до 18 років. Гіперпаратиреоз діагностується у 90% пацієнтів. У більшості хворих відзначається безсимптомна гіперкальціємія, в 25% випадків спостерігаються ознаки сечокам'яної хвороби. Наявність відповідних ДНК-зондів дозволяє виявити носіїв гена з 99% точністю при народженні, уникаючи непотрібних біохімічних скринінгових програм.
МЕН I виникає при мутації гена MEN1, який розташований на хромосомі 11q13, і кодує протеїн менін, що функціонує як супресор пухлин. Запропоновано гіпотезу двосторонньої рецесивної мутації, згідно з якою перша з них в домінантному алелі створює схильність до туморогенезу, а друга супроводжується елімінацією нормального алеля і демаскує дефектний алель, індукуючи розвиток пухлини.
Синдром МЕН IIа типу характеризується наявністю медулярної карциноми щитоподібної залози, феохромоцитоми (одиничної, білатеральної або множинної) і гіперпаратиреозу. Синдром був описаний Джоном Сіпплом у 1959 році. Він успадковується за аутосомно-домінантним типом з високим ступенем пенетрантності та експресивністю, що варіює.
В основі МЕН IIа лежать мутації у гені RET. Цей ген кодує білок, який бере участь у передачі сигналів у клітинах. Мутації у гені RET провокують зростання і поділ клітин за відсутності сигналів ззовні клітини. Це може привести до утворення пухлин в ендокринних залозах та інших тканинах.
Сімейна гіпокальціурична гіперкальціємія
Характеризується аутосомно-домінантним типом успадкування, нормальним рівнем ПТГ, помірною гіперкальціємією і гіпокальціурією. В основі захворювання лежать мутації гена CASR. Захворювання зазвичай протікає безсимптомно або проявляється неспецифічними симптомами у вигляді втоми, слабкості, артралгій, краніалгій.
Вторинний гіперпаратиреоз (Е 21.1)
Визначення
Вторинний гіперпаратиреоз – компенсаторна гіперфункція і гіперплазія прищитовидної залози, що розвивається при тривалій гіпокальціємії і гіперфосфатемії різного генезу.
Цей варіант гіперпаратиреозу найчастіше виникає у дитячому віці.
Етіологія
Причинами вторинного гіперпаратиреозу може бути:
- Ниркова патологія: хронічна ниркова недостатність, тубулопатії, нирковий рахіт.
- Кишкова патологія: синдром мальабсорбції.
- Кісткова патологія: остеомаляція, хвороба Педжета.
- Недостатність вітаміну D: захворювання нирок, захворювання печінки, спадкові ферментопатії.
- Злоякісні захворювання: мієломна хвороба.
Основними причинами вторинного гіперпаратиреозу є ниркова недостатність і хвороби системи травлення.
Патогенез
Гіперфосфатемія, обумовлена різними патофізіологічними факторами, зокрема, зменшенням маси функціонуючих нефронів, стимулює продукцію фактора росту фібробластів 23 (FGF-23) остеоцитам.
Свою дію FGF-23 реалізує через рецептор FGFR1, який функціонує лише у тому випадку, якщо він ко-експресований з трансмембранним протеїном Klotho у вигляді Klotho-FGF- рецепторного комплексу. У проксимальних канальцях нирок FGF-23, інгібуючи експресію натрій-фосфатного ко-транспортеров II типу (NaPi-2a і NaPi-2c), знижує реабсорбцію фосфатів. Тобто, FGF-23 до певного моменту підтримує нормальний рівень фосфатів у сироватці крові. Також FGF-23 зменшує синтез ПТГ. Однак FGF 23 інгібує активність 1-альфа-гідроксилази, опосередковуючи зниження рівня активного метаболіту вітаміну Д – кальцитріолу (1,25 (OH) 2D3). Зниження рівня кальцитріолу супроводжується дефіцитом порушення його рецептора (vitamin D receptor – VDR) у клітинах слизової оболонки кишечника, що призводить до зниження синтезу кальцій-зв'язуючих протеїнів, і в клітинах ПЩЗ, збільшуючи синтез мРНК ПТГ.
Дефіцит кальцітріолу зменшує всмоктування кальцію у кишечнику, що призводить до гіпокальціємії та розвитку остеомаляції. Гіпокальціємія стимулює продукцію ПТГ, що сприяє підсиленій кістковій реабсорбції та руйнуванню кістки. Гіперфосфатемія і збільшення продукції FGF-23 супроводжується зниженням експресії VDR і кальцій-чутливих рецепторів (calcium-sensing receptor – CaSR), у зв'язку з чим клітини ПЩЗ втрачають здатність адекватно реагувати на зміни концентрації кальцію та/або кальцитріолу.
При тривалому впливі високих концентрацій FGF-23 відбувається фосфорилювання екстрацелюлярної регульованої кінази 1/2 у клітинах ПЩЗ, що призводить до значного зниження експресії FGFR1 і білка Klotho. Зниження експресії Klotho-FGF-рецепторного комплексу розгальмовує продукцію ПТГ.
Таким чином, на ранніх стадіях розвитку вторинного гіперпаратиреозу гіперпродукція ПТГ переважно зумовлена порушенням вітамін Д-асоційованої регуляції обміну кальцію і функціонування CaSR, а в подальшому дефіцитом активності FGF-23-асоційованих сигнальних шляхів.
Тривала стимуляція ПТГ веде до гіперплазії ПЩЗ: спочатку до поліклональної гіперплазії, а потім до моноклональної вузлової гіперплазії. Ключовим фактором, який визначає проліферацію клітин і розвиток гіперплазії ПЩЗ, є зниження активності сигналів, асоційованих з CaSR. Зниження активності CaSR призводить до дефіциту активації фактора транскрипції Sp1 і, як наслідок, до дефіциту трансактивності VDR. Недостатність експресії CaSR і VDR обумовлює зниження чутливості клітин ПЩЗ до інгібуючого впливу кальцію і кальцитріолу на секрецію ПТГ і сприяє розвитку гіперплазії ПЩЗ.
Клініка
У клінічній картині вторинного гіперпаратиреозу домінують симптоми основного захворювання, частіше за все, хронічної ниркової недостатності. Специфічними симптомами гіперпаратиреозу є болі в кістках, слабкість у проксимальних відділах м'язів, артралгії. Можуть виникати спонтанні переломи і деформація скелета. Утворення позакісткових кальцинатів має різні клінічні прояви. При кальцифікації артерій можуть розвиватися ішемічні зміни. На руках і ногах можуть бути виявлені периартикулярні кальцинати. Кальцифікація кон'юнктиви і рогівки у сполученні з рецидивуючим кон'юнктивітом позначається як синдром «червоного ока».
Діагностика гіперпаратиреозу
Діагностика первинного гіперпаратиреозу ґрунтується на виявленні гіперкальціємії, зниженого рівня неорганічного фосфору, підвищеного рівня ПТГ в плазмі крові. Для діагностики первинного гіперпаратиреозу рекомендується проведення 3-кратного дослідження загального або іонізованого кальцію (більш інформативно) у сироватці крові з інтервалом 2–3 тижні. При первинному гіперпаратиреозі рівень кальцію підвищений, зазвичай >2,85 ммоль/л. Гіперкальціємія є більш вираженою у дітей з гіперплазією прищитоподібних залоз, але показники кальцію не перевищують значення 3,0 ммоль/л. Кількість іонізованого кальцію завжди значно збільшена, навіть якщо загальний рівень кальцію є граничним або злегка підвищеним. Рівень сироваткового фосфору і магнію значно зменшується. У сироватці крові підвищується рівень залишкового азоту та сечової кислоти. Рівень лужної фосфатази підвищується у 1,5–6 разів, але у дітей може бути нормальним. Сироватковий рівень ПТГ підвищений, корелює з рівнем кальцію, у той же час рівень концентрації кальцитоніну відповідає віковій нормі. Сеча може мати постійну низьку питому вагу (гіпоізостенурія).
Для вторинного гіперпаратиреозу характерні нормокальціємія або помірна гіпокальціємія в поєднанні з підвищеним рівнем паратгормону. Крім того, характерна гіперфосфатемія, високий рівень лужної фосфатази, низький рівень кальцитріолу. Визначення рівня паратгормону рекомендується при нефропатії будь-якого генезу зі зниженням швидкості клубочкової фільтрації менше 60%.
Для визначення об'ємних утворень або гіперплазії ПЩЗ використовують різні методи візуалізації: УЗД, сцинтіграфію, мультиспіральну комп'ютерну томографію (КТ). При малій масі ПЩЗ УЗД є малоінформативною процедурою. Більш ефективним методом візуалізації ПЩЗ є мультиспіральна КТ, яка дозволяє виявити аденоми розміром 0,2–0,3 см та їх атипову локалізацію. Для більш детальної топічної діагностики можливого розташування ОЩЖ проводиться сцинтиграфія з використанням технетріла 99mTc. Чутливість даного методу досягає 91%.
Для діагностики наявності остеопорозу проводять остеоденситометрію та рентгенологічне дослідження. Інформативною є остеоденситометрія поперекового відділу хребта, проксимального відділу стегнової, а також дистального відділу променевої кістки. Рентгенологічними ознаками первинного гіперпаратиреозу є дифузний остеопороз, витончення кортикального шару з розширенням кісково-мозкового каналу. Специфічною ознакою первинного гіперпаратиреозу є зменшення щільності кортикальної тканини в порівнянні з губчастою. Близько 10% хворих мають рентгенологічні ознаки рахіту. Кісткові зміни при вторинному гіперпаратиреозі схожі з первинним гіперпаратиреозом (остеопороз, субперіостальна і субхондральна резорбція кісток кисті та ін.).
Рентгенограма черевної порожнини може виявляти ниркові камені або нефрокальциноз. Наявність остеопорозу є показанням для дослідження рівнів маркерів кісткової резорбції і кісткоутворення. Маркерами кісткової резорбції є N- і C-телопептиди молекул колагену 1-го типу (NTX, CTX), тартратрезистентна кисла фосфатаза (TRACP), а маркерами кісткоутворення – остеокальцин, карбокси- і амінотермінальні про-пептиди проколагену 1-го типу (Р1СР, Р1NP), загальна лужна фосфатаза (ALP) та її кістковий ізофермент (bALP).
При вторинному гіперпаратиреозі необхідна діагностика основного захворювання (хронічна ниркова недостатність, мальабсорбція та ін.).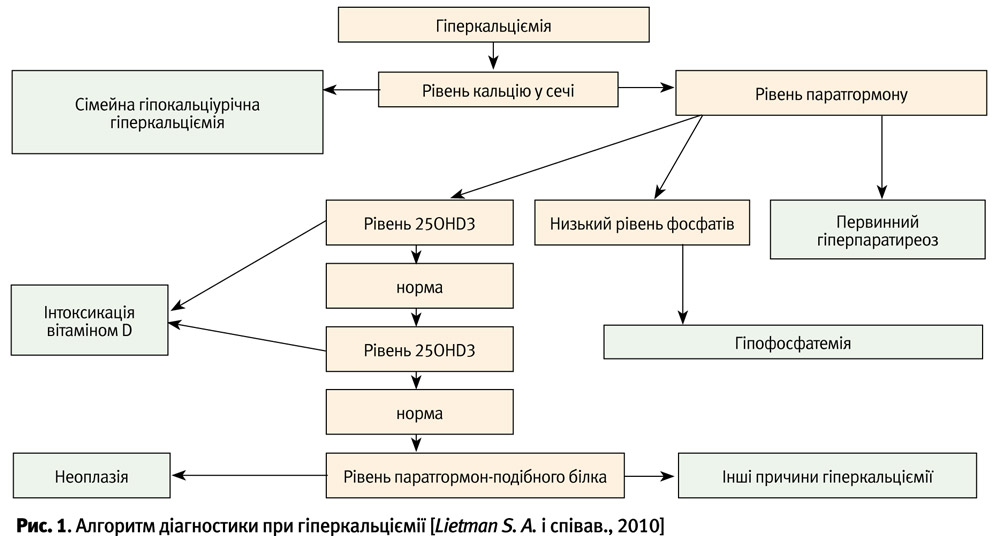
Диференційна діагностика первинного гіперпаратиреозу
Диференційна діагностика первинного гіперпаратиреозу проводиться із захворюваннями, що супроводжуються гіперкальціємією (рис. 1). Гіперкальціємія у дітей може спостерігатися при досить багатьох захворюваннях (табл. 1).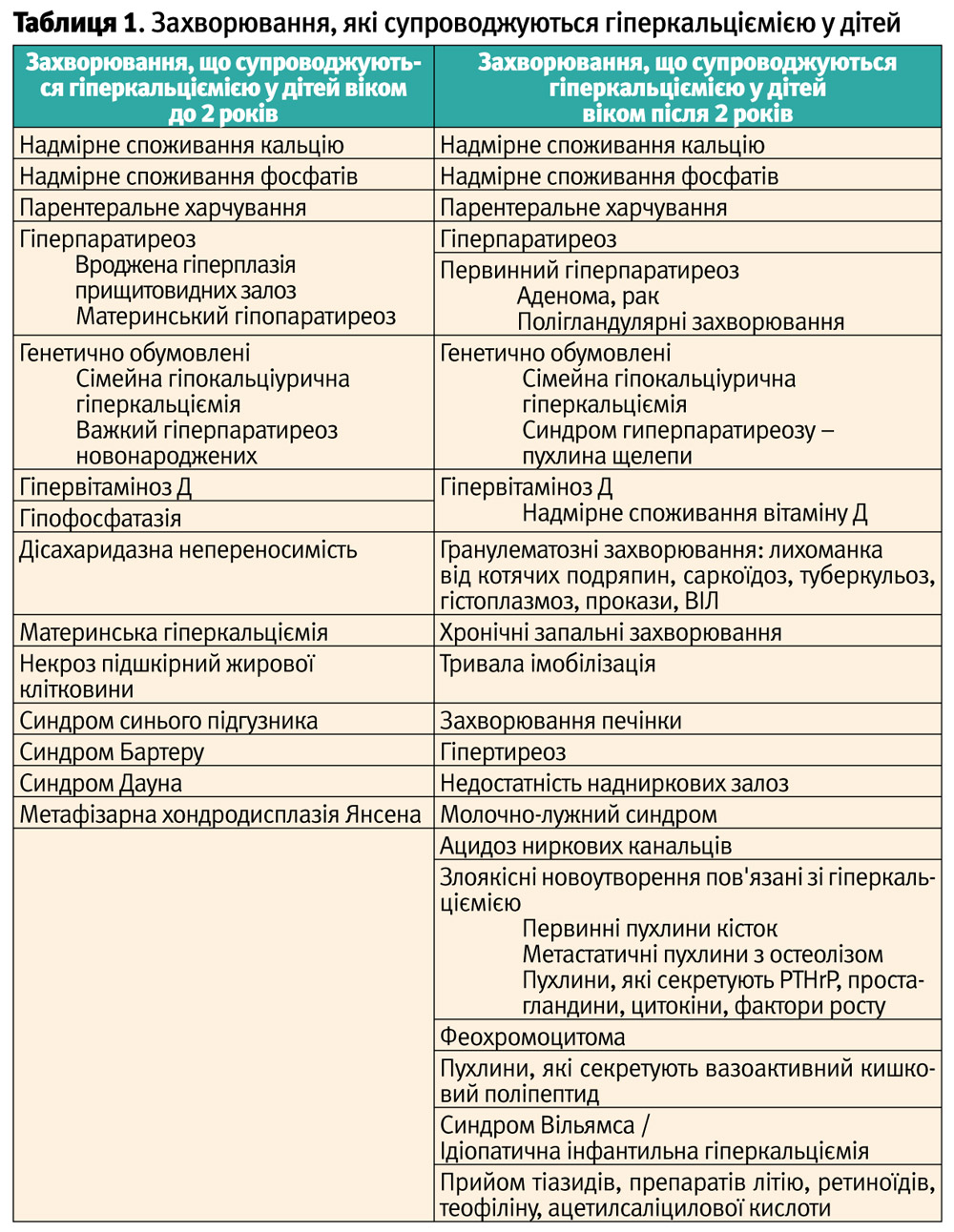
Лікування гіперпаратиреозу
При первинному гіперпаратиреозі практично у всіх випадках рекомендується оперативне лікування. Необхідне ретельне обстеження ПЩЗ. Якщо аденома виявлена, вона повинна бути вилучена. Більшість новонароджених з тяжкою гіперкальціємією потребують загальної паратиреоїдектомії, при легкій гіперкальціємії від хірургічного лікування можна утриматись. Після оперативного лікування пацієнт повинен отримувати лікування гіпопаратиреозу на протязі 6–12 місяців, потім рівні кальцію приходять до норми.
При вторинному гіперпаратиреозі показана профілактика остеопатії. З цією метою призначають активні препараті вітаміну D (альфа кальцидіол, кальцитріол). При триразовому підвищенні рівня паратгормону та/або підвищенні рівня кальцію крові більше 2,6–2,7 ммоль/л показана субтотальна паратиреоїдектомія.
Ефективність та безпека препаратів групи бісфосфонатів (алендронової, ібандронової, золедронової кислот) і кальціміметиків (цинакальцета) у дітей вивчені недостатньо.
Прогноз
Прогноз відносно сприятливий. Після успішного оперативного лікування більшість симптомів гіперпаратиреозу зникає протягом 6–12 місяців.
Перелік літератури знаходиться у редакції
Детальніше
МУКОВІСЦИДОЗ: прогрес у розумінні патогенезу, діагностики та лікування
 У старовинних трактатах та у фольклорних піснях, від часів середньовіччя, зустрічаються згадки про зв‘язок між «солоною шкірою» у дітей та їхньою смертністю у ранньому віці. Ці дані можна вважати першими описами найпоширенішого аутосомно-рецесивного захворювання осiб європейсько-азіатської популяцiї — муковісцидозу (МВ, Cуstic Fibrosis, CF). Перші описи захворювання, що засвідчували зв’язок між ущільненням підшлункової залози та порушенням функцій травної і дихальної систем, з’явилися наприкінці 30-х років XX-го століття: у 1936 р. Fanconi на педіатричному конгресі у Швейцарії вперше подав доповідь «Целіакія з бронхоектазією», а у 1938 р. Andersen на основі характеру змін підшлункової залози, що виникають при цьому захворюванні, дала йому назву «кістозний фіброз підшлункової залози». У подальшому звернули увагу на те, що у хворих на кістозний фіброз підшлункової залози надзвичайно в’язкий секрет утворюється і в інших залозах. Тому назва «муковісцидоз», утворена зі слів «mucus» (слиз) і «viscid» (густий, липкий), достатньо глибоко відображає процеси, які відбуваються при цьому захворюванні. У 1958 р. Gibson i Cook впровадили пілокарпіновий тест для визначення рівня хлоридів у потовій рідині, який і на сьогодні вважається «золотим стандартом» у діагностиці муковісцидозу. Від часу ідентифікації гена CFTR ( ТРБМ – Трансмембранний Регуляторний Білок Муковісцидозу), мутації якого спричинюють виникнення МВ, минуло понад 20 років, і вченими накопичено значну кількість відомостей про його структуру в нормі та при патології. Завдяки ранній діагностиці, більш активним методикам терапії, за останні кілька десятиліть медіана віку виживання хворих на МВ у розвинених країнах перевищила 30 років.
У старовинних трактатах та у фольклорних піснях, від часів середньовіччя, зустрічаються згадки про зв‘язок між «солоною шкірою» у дітей та їхньою смертністю у ранньому віці. Ці дані можна вважати першими описами найпоширенішого аутосомно-рецесивного захворювання осiб європейсько-азіатської популяцiї — муковісцидозу (МВ, Cуstic Fibrosis, CF). Перші описи захворювання, що засвідчували зв’язок між ущільненням підшлункової залози та порушенням функцій травної і дихальної систем, з’явилися наприкінці 30-х років XX-го століття: у 1936 р. Fanconi на педіатричному конгресі у Швейцарії вперше подав доповідь «Целіакія з бронхоектазією», а у 1938 р. Andersen на основі характеру змін підшлункової залози, що виникають при цьому захворюванні, дала йому назву «кістозний фіброз підшлункової залози». У подальшому звернули увагу на те, що у хворих на кістозний фіброз підшлункової залози надзвичайно в’язкий секрет утворюється і в інших залозах. Тому назва «муковісцидоз», утворена зі слів «mucus» (слиз) і «viscid» (густий, липкий), достатньо глибоко відображає процеси, які відбуваються при цьому захворюванні. У 1958 р. Gibson i Cook впровадили пілокарпіновий тест для визначення рівня хлоридів у потовій рідині, який і на сьогодні вважається «золотим стандартом» у діагностиці муковісцидозу. Від часу ідентифікації гена CFTR ( ТРБМ – Трансмембранний Регуляторний Білок Муковісцидозу), мутації якого спричинюють виникнення МВ, минуло понад 20 років, і вченими накопичено значну кількість відомостей про його структуру в нормі та при патології. Завдяки ранній діагностиці, більш активним методикам терапії, за останні кілька десятиліть медіана віку виживання хворих на МВ у розвинених країнах перевищила 30 років.
У розвинених країнах в останні роки відзначається зростання числа хворих на МВ підліткового, юнацького віку і дорослих, що свідчить про поступову його трансформацію з фатального захворювання дитячого віку в хронічну патологію дорослих. Така трансформація великою мірою зумовлена постійним поглибленням уявлень про патогенез муковісцидозу та поширенням цих знань серед фахівців, що стикаються із такими пацієнтами.
Ген ТРБМ та його мутації
Патогенез муковісцидозу зумовлений порушенням ТРБМ – АТФ-зв‘язаного транспортного білка, що виконує функцію каналу аніонів хлору в апікальних клітинах залоз зовнішньої секреції. ТРБМ присутній у високій концентрації на апікальних мембранах клітин бронхів, слинних залоз, підшлункової залози, залоз кишківника та епітеліальних клітинах сім’явивідних шляхів. При МВ секрети екзокринних залоз густішають, що приводить до розвитку мультисистемного захворювання (з ураженням бронхо-легеневої системи, системи травлення, у першу чергу підшлункової залози і печінки, репродуктивної системи). Ураження лише однієї із систем органів веде до розвитку ізольованих форм МВ, до яких, зокрема, належить і генітальна форма МВ.
На сьогодні виявлено більше 1900 мутацій гена ТРБМ, відповідальних за розвиток симптомів MB, з яких більшість є рідкісними або навіть унікальними. Частота та спектр цих мутацій варіює в залежності від географічного положення та етнічного складу вибірки. Мутації гена ТРБМ виявляють як при класичному МВ, так і при інших фенотипічних проявах, пов'язаних з мутаціями гена ТРБМ (CFTR-relateddisorders – CFTR-RD). До них, зокрема, належать дисеміновані бронхоектази, хронічний панкреатит, хронічний синусит, а також вроджена аплазія сім’явивідних протоків. Для усіх вищенаведених нозологій генетичне тестування мутацій гена CFTR є обов’язковою складовою їх діагностичного алгоритму.
Мутації гена CFTR можна розділити на п'ять або шість загальних класів відповідно до їх впливу на синтез ТРБМ і порушення функції каналу хлору. Такий поділ став основою різних підходів до розроблення патогенетичної терапії. У світі відбувається уже ІІ–ІІІ фази клінічних випробувань нових засобів для корекції біохімічного дефекту при МВ. Тому молекулярно–генетичне дослідження мутацій гена ТРБМ необхідне не тільки для пре- та постнатальної діагностики МВ, виявлення здорових гетерозиготних носіїв, але й визначення правильних підходів до лікування МВ.
Ідентифіковано групу мутацій, що асоціюють звичайно зі збереженням функції підшлункової залози, і визначають, як «м'які» щодо статусу підшлункової залози. «М'які» мутації мають домінантний ефект, тому що вони асоційовані із залишковою екзокринною функцією підшлункової залози навіть у пацієнтів, що несуть важку мутацію в другому aлелі. До таких алелів, поширених в Україні, належать мутації 3849+10kbC>T та R334W.
Найбільш поширеною мутацією в усіх популяціях є делеція трьох нуклеотидів у 10 екзоні, що призводить до втрати залишку фенілаланіну в 508 положенні молекули білка (F508del). Відносна частка цієї мутації становить близько 66% усіх МВ хромосом, обстежених у світі, близько 45% усіх хворих на МВ є гомозиготами за мутацією F508del. Другою за поширенням мутацією гена ТРБМ в Україні є мутація 2184insA. Її частота є найвищою у Західній Україні (понад 7,2% усіх мутантних хромосом), і припускається, що ця мутація виникла у слов’янських популяціях Галичини, звідки розповсюдилася в Європу та за її межі.
Діагностика муковісцидозу
Діагноз МВ базується на наявності хронічного бронхолегеневого процесу, кишкового синдрому, позитивного потового тесту, ідентифікації мутацій у гені ТРБМ, обтяженого сімейного анамнезу. Від 2012 року в Україні запроваджено неонатальний скринінг на МВ на основі двокрокового вимірювання рівня імунореактивного трипсиногену, метою якого є ідентифікація хворих на МВ на максимально ранніх, часто доклінічних, стадіях захворювання. Проте, як і інші скринінгові програми, ця програма також вимагає етапу верифікації діагнозу «муковісцидоз».
Нові критерії діагностики МВ включають два блоки:
- Один із характерних клінічних симптомів або випадок МВ у сім’ї, або позитивний результат неонатального скринінгу за імунореактивним трипсином.
- Підвищена концентрація хлоридів у потовій рідині або дві ідентифіковані мутації гена ТРБМ, або позитивний тест різниці назальних потенціалів.
Діагноз вважається достовірним, якщо наявні хоча б по одному критерію з кожного блоку.
Класифікація МВ включає: МВ з панкреатичною недостатністю; МВ без панкреатичної недостатності та ТРБМ-зумовлені фенотипи.
Наведені формулювання діагностичних критеріїв свідчать про вагомість параклінічних показників (потова проба, генетичне дослідження та визначення зовнішньої секреторної функції підшлункової залози) для встановлення діагнозу МВ.
Потова проба повинна проводитися у всіх випадках підозри на дане спадкове захворювання. Позитивним вважають потовий тест при встановленні концентрації іонів хлору понад 60 мекв/л, сумнівним – при 40–60 мекв/л, негативним – менше 40 мекв/л. Діагностично значимим для МВ вважають отримання позитивного результату двох або більше аналізів поту. Для дітей менше одного року значення концентрації електролітів у поті більше 40 мекв/л може бути показником для встановлення діагнозу «муковісцидоз». Використання для аналізу недостатньої кількості поту призводить до отримання псевдопозитивних результатів. Важливість результатів потової проби для підтвердження чи заперечення діагнозу МВ вимагає значної уваги до її проведення.
Оскільки муковісцидоз – генетичне захворювання, для його діагностики є необхідним проведення аналізу ДНК. Суть даного дослідження полягає у виявленні у конкретного пацієнта змін у структурі ДНК гена ТРБМ. Молекулярно-генетична діагностика мутацій гена ТРБМ повинна проводитися: у пацієнтів із позитивним результатом потового тесту; у пацієнтів із клінічними проявами МВ; в усіх членів родин, де виявлено випадки захворювання на МВ; при підозрі на атипову форму МВ, наприклад, у чоловіків із порушеними показниками спермограми нез’ясованого ґенезу; у вагітних і сімейних пар без сімейного анамнезу, що повідомлені про ризик МВ і хочуть провести ДНК-діагностику.
Матеріалом для молекулярно-генетичного аналізу є ДНК, яка може бути виділеною із будь-яких ядерних клітин організму. Проте, у практиці використовують лейкоцити периферичної крові і для проведення ДНК-діагностики потрібний забір 2–4 мл венозної крові у пробірку з ЕДТА. Забір крові можна проводити незалежно від прийому їжі чи медикаментів, обмеженням є процедура переливання крові чи її компонентів, після якої повинно минути не менше 14 днів. Проведення ДНК-діагностики є трудоємним дослідженням, що вимагає високої кваліфікації у виконавців та особливого вартісного обладнання. У зв‘язку із цим, даний аналіз, як і в інших країнах, в Україні проводиться лише у декількох наукових центрах, одним із яких є ДУ «Інститут спадкової патології НАМНУ».
Інтерпретація результатів генетичного дослідження
Якщо внаслідок генетичного тестування було виявлено мутації у двох алелях гена ТРБМ (гомозиготи або компаунд гетерозиготи), діагноз «муковісцидоз» є повністю підтвердженим. Найчастіше виявляється мутація F508del. Пацієнтів, у яких у двох алелях є дана мутація, називають гомозиготами за мутацією F508del й генотип записують F508del/F508del. Можливі варіанти, коли виявлено також іншу мутацію, окрім F508del. У цьому випадку у результаті генетичного тестування записано, що пробанд є компаундною гетерозиготою за певними мутаціями. Наприклад, генотип F508del/2184insA. Виявлені у пацієнта мутації повинні бути ідентифікованіі у його батьків.
Ідентифікація однієї мутації у пробанда із клінічними проявами МВ також дає всі підстави підтвердити муковісцидоз. Якщо у пацієнта виявлено мутацією лише в одному алелі, то це означає, що друга мутація не була ідентифікованою. Часом такі результати можна сплутати із гетерозиготним носійством мутацій гена ТРБМ, при якому у пацієнтів немає проявів МВ.
Негативний результат генетичного аналізу («Не виявлено жодної із досліджуваних мутацій гена ТРБМ») вказує лише на відсутність у даного індивіда досліджуваних мутацій, а не заперечує діагноз «муковісцидоз». Хоча при цьому ризик МВ зменшується залежно від кількості протестованих мутацій. З кожним роком зростають діагностичні можливості лабораторій, і наявність зразка ДНК дозволяє проводити у пацієнта додаткові обстеження і встановити тип мутацій через досить тривалий час після здачі крові.
Мутації гена ТРБМ, що були ідентифіковані у хворого на муковісцидоз (пробанд), виявляються і у членів його родини. Найперше потрібно звертати увагу на сибсів (рідних братів і сестер), а також близьких родичів репродуктивного віку.
Профілактика та дородова діагностика муковісцидозу
Для сімей, у яких є діти, хворі на МВ, існує підвищений ризик повторного народження хворої дитини: 25% для кожної наступної вагітності. Значення можливості народити здорову дитини для таких сімей важко перебільшити. Батьки дітей, у яких верифіковано генетично детерміновані захворювання, в основному уникають повторних вагітностей, що мотивується небажанням мати ще одну хвору дитину та погіршенням можливостей забезпечити їй необхідне лікування та догляд. Незважаючи на значний прогрес у терапії МВ, значному продовженні тривалості та покращенні якості життя таких пацієнтів, лікування вимагає від родини значних матеріальних, моральних та фізичних затрат. У випадку пренатальної діагностики МВ матеріалом для дослідження є ДНК плода, виділена з клітин хоріона або навколоплідних вод. Також можливою є доімплантаційна діагностика МВ у випадку застосування екстракорпорального запліднення. Забір матеріалу для пренатальної діагностики проводиться за допомогою інвазивних методів, таких як біопсія хоріону або плаценти (8–12 тиждень вагітності) чи амніоцентез (забір навколоплідних вод на 16–20 тиждень вагітності). Забір матеріалу проводиться у гінекологічних стаціонарах. Матеріал для генетичного аналізу пересилається у лабораторії, у яких проводилася ДНК-діагностика мутацій гена ТРБМ у пробанда та батьків. Важливим є повідомлення про інформативну суть даного дослідження, що полягає лише у наданні інформації щодо стану плоду, а рішення щодо пролонгування чи переривання вагітності приймають батьки. У результаті 20 пренатальних діагностик МВ, проведених в ДУ «ІСП НАМНУ», у 15 (75%) випадках у плодів виявлено відсутність мутацій гена ТРБМ (у гомо- (35%) чи гетерозиготному (40%) станах) та народилися здорові діти у сім’ях, де зареєстровані випадки МВ. Зважаючи на високу частоту гетерозиготного носійства мутацій гена ТРБМ у популяції, при плануванні вагітності носіями МВ обов’язковим є тестування партнера.
Чоловіки, хворі на МВ, мають обструктивну азооспермію і у 95% випадків неплідні. Проте, запліднення може відбутися при застосуванні допоміжних репродуктивних технологій, оскільки клітини сперматогенезу таких чоловіків зберігають здатність до запліднення і можуть бути отримані шляхом біопсії яєчка. В осіб жіночої статі, хворих на МВ, збережена нормальна репродуктивна функція і основною проблемою є стан соматичного здоров’я жінки при вагітності та пологах. При відсутності мутацій гена ТРБМ у партнера ризик народження дитини, хворої на МВ, у такого подружжя є низьким. Народження дітей вагітними, хворими на МВ, є показником значного прогресу у лікуванні даного захворювання в Україні.
Терапевтичний супровід та моніторинг хворих на муковісцидоз
Згідно міжнародного досвіду, лікування та спостереження за хворими на МВ повинно відбуватися у спеціалізованих медичних центрах. Така вимога зумовлена необхідністю роботи команди фахівців (педіатра, пульмонолога, генетика, гастроентеролога, дієтолога, фізіотерапевта, рентгенолога, психолога, кваліфікованих медичних сестер та соціальних працівників), яка зможе забезпечити вирішення різних проблем, з якими стикаються хворі на МВ. Гострою проблемою для України є функціонування дорослих центрів МВ, оскільки за минулу декаду в Україні значно зросла і далі зростатиме кількість хворих на МВ віком більше 18 років. У світі є різний досвід організації таких центрів. У деяких країнах є центри МВ, де відбувається спостереження і лікування хворих на МВ різного віку, але у багатьох країнах центри МВ для дорослих функціонують окремо. У центрах МВ для дорослих додатково працюють андрологи, гінекологи, ендокринологи, а також є зв'язок із клініками, де відбувається трансплантація органів, оскільки значна кількість пацієнтів старшого віку підлягає трансплантації легень.
Зважаючи на мультисистемність уражень при МВ, для таких пацієнтів слід одночасно вирішувати багато клінічних проблем, які будуть коротко розглянуті нижче.
Зовнішньо-секреторна недостатність підшлункової залози. Екзокринна недостатність підшлункової залози зустрічається у 85–90% хворих на МВ. Рівень зовнішньо-секреторної недостатності підшлункової залози вимірюють шляхом визначення рівня фекальної елестази-1, значення якого у хворих на МВ може знижуватися з віком.
Через брак панкреатичних ферментів у травному тракті виникають симптоми панкреатичної недостатності, клінічними ознаками якої є збільшений у розмірах живіт, абдомінальні болі, поліфагія, поліфекалія, зміна випорожнень. До симптомів мальдигестії швидко приєднується вторинний синдром мальабсорбції. Найчастішою проблемою з боку тонкої кишки, зумовленою зовнішньо-секреторною недостатністю підшлункової залози у новонароджених, є меконіальний ілеус. У 15–20% дітей старшого віку, хворих на МВ, еквівалентом меконіального ілеусу є синдром дистальної інтестінальної обструкції (СІДО). Для компенсації зовнішньо-секреторної недостатності підшлункової залози усі хворі на МВ вимагають пожиттєвого призначення замісної терапії панкреатичними ферментами в індивідуально підібраних дозах.
Особливості терапії бронхо-легеневих проблем. Відомо, що причиною несприятливого прогнозу хвороби у 95% хворих МВ є бронхо-легенева патологія. Для ліквідації загострення бронхо-легеневого процесу тактика не змінилася – використання тривалої високодозної двокомпонентної довенної антибіотикотерапії. Слід відмітити, що хворі на МВ мають особливості фармакокінетики антибіотиків, які обґрунтовують вищеперераховані підходи до антибактеріальної терапії. Їм притаманне збільшення системного кліренсу, пришвидшення метаболізму в печінці і збільшення ниркового кліренсу. Крім того, максимальна концентрація антибіотиків у сироватці крові у хворих на МВ є нижчою, ніж при введенні тієї ж дози препарату хворим з іншою патологією. Внутрішньо-бронхіальне розташування мікроорганізмів, погане проникнення в харкотиння більшості антибіотиків, антибіотикостійкість бактерій, що часто зустрічається у хворих на МВ, зумовлює необхідність тривалої довенної двокомпонентної високодозної антибіотикотерапії, а також поєднане застосування їх інгаляційних форм.
Вибір антибіотика в хворих на МВ визначається видом мікроорганізмів, що виділяються з бронхіального секрету, і їх чутливістю до антибіотиків. Найбільш розповсюдженими є St. aureus, H. influenzae і P. aeruginosa (мукоїдна і немукоїдна форма). Останнім часом зросла роль Burkholderia cepacia, що характеризується полірезистентністю до антибіотиків. Контроль інфекційного синдрому дозволяє утримувати в компенсованому стані енергетичний баланс і попереджувати погіршення нутрітивного статусу при даній патології.
Аналізуючи клінічні випадки хворих на МВ, яким давали антибіотики в середньотерапевтичних дозах, спостерігалися часті рецидиви хронічного інфекційного процесу в бронхах, «культивація» нечутливих до антибіотиків штамів патогенів, швидке формування незворотніх структурних змін – множинних бронхоектазів і пневмофіброзу, а, відтак, погіршення прогнозу захворювання.
Слід наголосити на одному з високоефективних і малозатратних компонентів терапії бронхо-легеневого процесу при МВ — кінезітерапії, основна мета якої є очищення бронхіального дерева від в’язкого харкотиння. Широко використовується перкусія і вібрація грудної клітки, активний цикл дихання і аутогенний дренаж. Високоефективними і малозатратними в терапії МВ є інгаляції з концентрованими розчинами NaCl та бромгексином.
З метою запобігання прогресуванню гепатобіліарних проблем хворі на МВ отримують урсодеоксихолеву кислоту (УДХК), яка має пряму цитопротективну дію, підвищує печінково-кишкову рециркуляцію, збільшує пасаж жовчі і виділення жовчних кислот через кишку, зменшує літогеність жовчі. Гастроезофагеальний рефлюкс (ГЕР) значною мірою погіршує якість життя хворих на МВ. На фоні комплексної симптоматичної терапії ГЕР покращується апетит, регресує абдомінальний больовий синдром, знижується доза панкреатичних ферментів та навіть частота рецидивів бронхо-обструктивного синдрому.
Крім енергетичного дефіциту, у хворих на МВ закономірно розвивається дефіцит жиророзчинних вітамінів (А, D, Е, K), незамінних жирних кислот, а також деяких мікроелементів, що вимагає їх введення у план базової терапії. Дуже важливим є правильний розрахунок необхідних додаткових калорій, що покриватимуть підвищенні енергетичні затрати організму хворого на МВ.
Вагомий вплив на перебіг МВ і, відповідно, якість життя хворих на МВ має ретельність виконання лікарських призначень і план моніторного спостереження пацієнтів. Оцінка антропометричних даних, визначення насичення киснем периферійної крові, посів харкотиння слугують інформативними і неінвазивними критеріями для встановлення ступеня компенсації МВ в цілому. При виявлені відхилень у нутрітивному статусі проводиться пошук причини відхилень та проводиться відповідна корекція терапевтичного супроводу. При неефективності терапії, проведеної в амбулаторних умовах, показане поглиблене обстеження і лікування в умовах стаціонару.
Максимальна ефективність протокольної терапії МВ, яка не тільки позитивно впливає на стан хворого, але й покращує прогноз хвороби, продовжує тривалість життя із задовільною якістю, можлива тільки тоді, коли діагностика і призначення лікування співпадає з дебютом перших проявів хвороби і, відповідно, хвороба перебуває на стадії, коли ще немає незворотніх змін з боку життєво важливих органів і систем.
Література
- Л. Й. Бобер. Стан підшлункової залози у хворих на муковісцидоз залежно від асоціації фенотип–генотип.: автореферат дис. на здоб. вченого ступеня к. мед. н.: спец. 14.01.10. «Педіатрія». Львів–2009.
- Горовенко Н. Г. Узгоджені рекомендації щодо діагностики, лікування і профілактики муковісцидозу // Укр. пульмонологічний журнал. –1999, N1. – c.14-17.
- Лівшиць Л. А. Природа, походження та шляхи розповсюдження мутацій, що спричинюють моногенні спадкові захворювання: Автореф. на здоб. вченого ступеня д. біол. н. – Київ. – 2001.
- Методики молекулярно-генетичного аналізу муковісцидозу, синдрому Ніймеген та фенілкетонурії // Методичні рекомендації. – Укл. Макух Г. В., Гнатейко О. З., Заставна Д. В. та ін. – Київ. – 2009. – 28 c.
- Петрова Н. В. Молекулярно-генетические и клинико-генотипические особенности муковисцидоза в российских популяциях: автореф. дис. на соискание ученой степени д. биол. н.: спец. 03.00.15. «Генетика» / Н. В. Петрова. – M., 2009.
- A high frequency of the Cystic Fibrosis 2184insA mutationin Western Ukraine: Genotype-phenotype correlations, relevance for newborn screening and genetic testing/ H. Makukh, P. Krenkova, M. Tyrkusetal. // Journal of Cystic Fibrosis, – 2010, N9, – P. 371-375.
- Best practice guidelines for molecular genetic diagnostics of cystic fibrosis and CFTR- related disorders – updated European recommendations / Els. Dequeker, M. Stuhrmann, M. Morisetal. // European Journal of Human Genetics, – 2008. – N3, – P. 1-15.
- Čamajova J. Variability in the use of CE-marked assays for in vitro diagnostics of CFTR gene mutations in European genetic testing laboratories /Čamajova J., Berwouts S., Matthijs G., MacekJr M., Dequeker E. // Eur. J. Hum. Genet. – 2009; N17. – P. 537-540.
- CFTR mutations in men with congenital bilateral absence of the vasdeferens (CBAVD): a systemic review and meta-analysis / J. Yu, Z. Chen, Y. Ni, Z. Li // Hum Reprod. – 2012. – Vol. 27 (1). – P. 25-35.
- Consensus on the use and interpretatation of cystic fibrosis mutation analysis in clinical practice / C. Castellani, H. Сuppens, M. Macek [etal.] // Journal of Cystic Fibrosis. – 2008. – Vol.7. – P. 179-196.
- Cystic Fibrosis Mutation DataBase: http://www.genet.sickkids.on.ca/cftr.
- European best practice guidelines for cystic fibrosis neonatal screening /Castellani C., Southern. K. W., Brownlee K. et al // J. Cyst. Fibrosis. – 2009; Vol. 3, N 8, – p.153-173.
- Frequency and origin of 2184insA mutationin CF patients from Ukraine / S. A. Kravchenko, H. V. Makukh, M. Pampukha [etal.] // Europ. J. Hum. Genet. – 2011. – Vol. 19, Supl 2. – P. 332.
- Guidelines for diagnosis of cystic fibrosis in newborns throug holder adults: Cystic Fibrosis Foundation consensus report / P. M. Farrell, B. J. Rosenstein, T. B. White [etal.]; Cystic Fibrosis Foundation // J. Pediatr. – 2008. – Vol.153 (2). – P. S4-S14.
- New clinical diagnostic procedures for cystic fibrosis in Europe / K. De Boeck, N. Derichs, I. Fajac [etal.]; ECFS Diagnostic Network Working Group; Euro Care CF WP3 Group on CF diagnosis // J. Cyst Fibros. – 2011. – Vol. 10 (Suppl 2). – P. S53-S66.
- Phenotypic and genetic characterization of patients with features of «nonclassic» forms of cystic fibrosis / J. D. Groman, B. Karczeski, M. Sheridan [etal.] // J. Pediatr. – 2005. – Vol. 146. – P. 675-680.
- Proesmans M. What's new in cystic fibrosis? From treating symptoms to correction of the basic defect / M. Proesmans, F. Vermeulen, K. De Boeck // Eur J. Pediatr. – 2008. – Vol. 167 (8). – P. 839-849
- Recommendations for the classification of diseasesas CFTR-related disorders / C. Bombieri, M. Claustres, K. De Boeck [etal.] // J Cyst Fibros. – 2011. – Vol. 10 (Suppl 2). – P. S86-S102.
- Rogan M.P. Cystic fibrosis transmembrane conduct an ceregulator intracellular processing, trafficking, and opportunities for mutation-specific treatment / M. P. Rogan, D. A. Stoltz, D. B. Hornick // Chest. – 2011. – Vol. 139 (6). – P. 1480-1490.
- Rosenstein B. J., Cutting G. R. The diagnosis of cystic fibrosis: a consensus statement. Cystic Fibrosis Foundation Consensus Panel // Journal of Pediatrics. –1998. – Vol. 132, N4. – P. 589-595.
- Standard of care for patients with Cystic Fibrosis: A European Consensus. // Journal of Cystic Fibrosis, – N4, –2005, – P. 7-26.
- Sultan M. Genetic prevalence and characteristics in children with recurrent pancreatitis / M. Sultan, S. Werlin, N. Venkatasubramani // J Pediatr Gastroenterol Nutr. – 2012. – Vol. 54 (5). – P. 645-650.
- Tsui L.C. The cystic fibrosis transmembrane conduct an ceregulator gene / L. C. Tsui // American Journal of Respiratory&Critical Care Medicine. – 1995. – Vol. 151(3). – P. 47-53.
- Wilschanski M. New drugs for cystic fibrosis / M. Wilschanski, E. Kerem // Expert Opin. Investig. Drugs. – 2011. – Vol. 20 (9). – P. 1285-1292.

ШЛЯХ ДОБРА
Святий Миколай завітав до дітей в макіївський дитбудинок «Проліски». 55 дітей віком від 6 до 15 років отримали книжки, постільну білизну, побутову хімію, канцелярію та ще багато необхідних речей
Місяць тому волонтери через соціальні мережі поширили заклик до збору подарунків.
Багато ЗМІ підтримали акцію і розповіли про неї своїм читачам та глядачам. Так, протягом чотирьох тижнів небайдужі кияни зносили свої дари до «торбинки Святого Миколая». Три величезні святкові торби, а також дві коробки необхідних речей, були доставлені активістами до Макіївки.
Суддя Вікторія Джарти, чия родина традиційно підтримує дитбудинок «Проліски», взяла активну участь в акції. Вона з’ясувала потреби дитячого будинку і долучилася до збору та закупівлі необхідних речей. Зокрема, старшокласники отримали від Вікторії інтелектуальні подарунки – електронні книжки. «Не переставайте мріяти, вірте в чудо, читайте та наполегливо працюйте – зазначила пані Вікторія, звернувшись до вихованців, – ці книжки можуть подарувати вам і вірних друзів – героїв творів, і дозволити подорожувати в часі та просторі; вони можуть дати вам життєвий досвід та підказати, як діяти у тій чи іншій ситуації».
Інна Бойко, представник організації пацієнтів ЮКАБ, яка доклала надзусиль для здійснення акцїї, підкреслила, що часточка свята та родинного тепла є в подарунках та іменних листівках, які всі охочі могли підписати кожній дитині особисто. «Для нас важливо дбати про тих, хто цього потребує, так само, як мобілізовувати суспільство у вирішенні соціальних проблем. Важлива участь кожної людини, адже для того, щоб допомагати ближньому, не треба бути олігархом. Хтось приніс пару шкарпеток, а хтось три книжки. Кожен зробив те, що може, але головне, що він не залишився осторонь».
Діти, що опинилися без батьківської опіки, були раді спілкуванню з гостями. Найшвидше вони розібрали саме іменні листівки, які були підписані усіма охочими для кожної дитини поіменно. Кожна дитина цього дня прочитала своє ім’я та теплі слова до свята на листівці. Можливо, саме їх вони збережуть на згадку, так само, як і волонтери збережуть бажання змінювати життя за допомогою маленьких, але важливих справ.
Детальніше
ДИАРЕЯ - диагностика причин
 Пищеварительный (желудочно-кишечный) тракт теплокровных животных и человека представляет собой систему органов, предназначенную для переработки и извлечения из пищи нутриентов (субстрата энергии), их всасывания в кровь, и обязательного выделения из организма неусвоенных остатков метаболизма. Таким образом, нарушение моторики, в частности, диарея (понос), является атрибутивным, и в тоже время крайне неспецифическим признаком патологии пищеварительной системы
Пищеварительный (желудочно-кишечный) тракт теплокровных животных и человека представляет собой систему органов, предназначенную для переработки и извлечения из пищи нутриентов (субстрата энергии), их всасывания в кровь, и обязательного выделения из организма неусвоенных остатков метаболизма. Таким образом, нарушение моторики, в частности, диарея (понос), является атрибутивным, и в тоже время крайне неспецифическим признаком патологии пищеварительной системы
«То, что для одного понос, для другого может быть запором» (английская поговорка)
Анатомия пищеварительного тракта человека
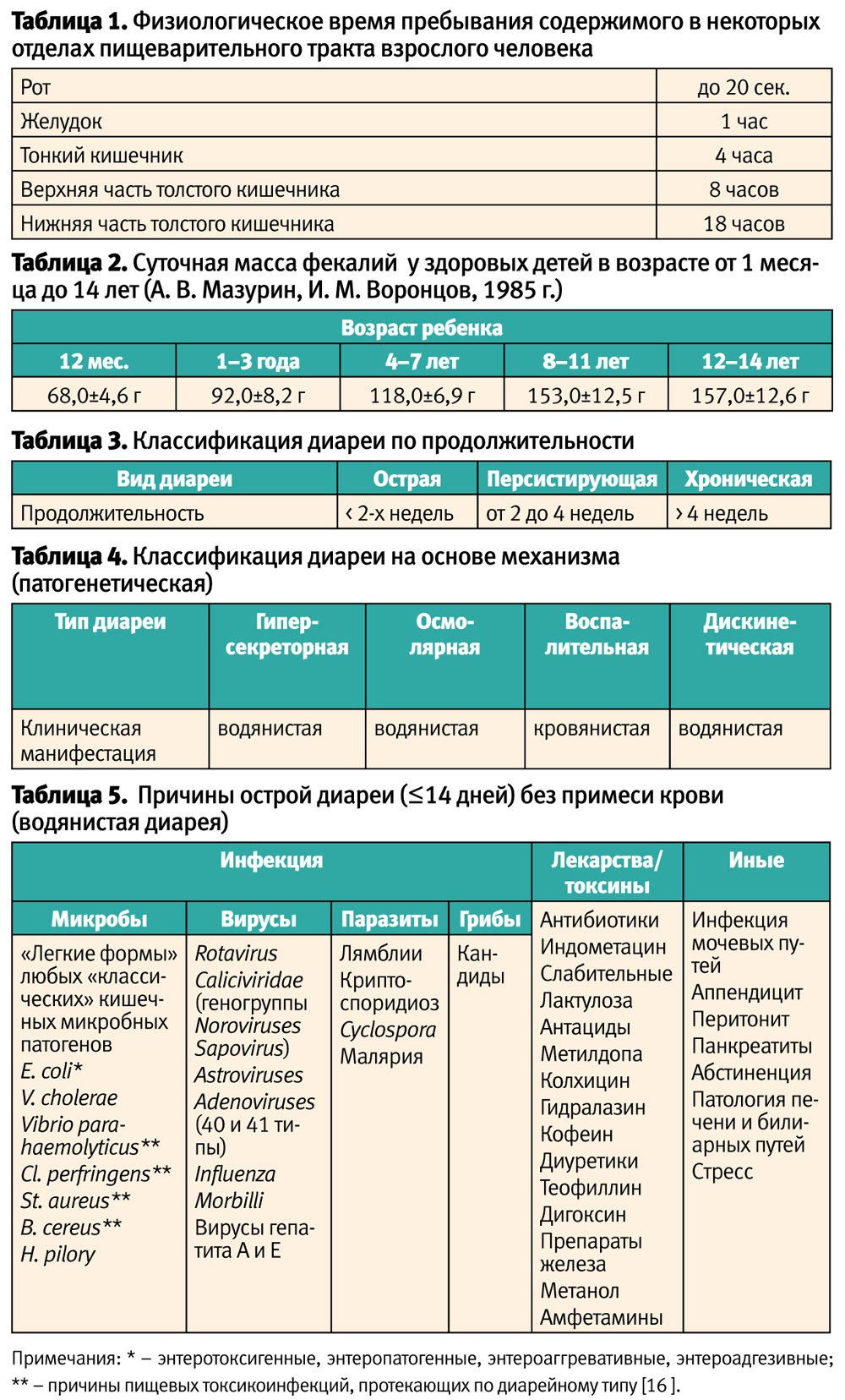
Пищеварительная система имеет чрезвычайно сложное анатомо-физиологическое устройство и включает не только мягкие ткани, но и костные образования, которыми являются зубы, или генетически отличающиеся биологические объекты в виде микрофлоры кишечника. Ферментация пищи и усвоение нутриентов в каждом отделе желудочно-кишечного тракта (ЖКТ) человека различно по времени, и в целом является относительно продолжительным процессом (табл. 1).
Дефиниция
Диарея, понос (от лат. diarrhea) – патологическая частота и консистенция испражнений [1]. В более строгом определении, которое не учитывает возрастные, гендерные и этно-географические особенности индивидуума, считается, что у пациента имеет место диарея, когда объем стула превышает 150 мл/сут [2]. В целом, для взрослого мужчины со стандартной массой тела объем каловых масс не должен превышать 225 мл/сут, а для женщин – 175 мл/сут [3]. При этом указанные стандарты соответствуют «западному» типу питания, при котором углеводы, как источник энергии, составляют не более 50–60% от общей калорийности пищевого рациона. В некоторых странах Африки и Азии доля углеводов в обеспечении организма энергией составляет 80%! [4]. Понятно, что при таком рационе преобладает растительная пища, богатая волокнами, что значительно изменяет консистенцию и объем испражнений.
В педиатрической практике понятие «диарея» применяется к детям, в том числе и грудного возраста, у которых объем испражнений превышает 10 мл/кг/сут [5]. По определению ВОЗ, диарея «представляет собой экскрецию необычно жидкого и водянистого кала, как правило, не менее трех раз в течение 24 часов. Однако особое значение в этом случае имеет не кратность стула, а его консистенция. Частая экскреция сформировавшихся каловых масс не указывает на наличие диареи. У детей, находящихся исключительно на грудном вскармливании, нередко бывает жидкий, «кашицеобразный» стул, это тоже не диарея» [6]. Следует помнить, что у детей в ЖКТ в общем обращается ≈285 мл/кг/сут жидкости. При этом испражнения у детей включают примерно (в расчете на литр): ≤25 мЕк натрия, ≤70 мЕк калия, ≤25 мЕк хлора. Средний объем каловых масс у ребенка в зависимости от возраста представлен в табл. 2 [7].
Таким образом, необходимо обратить внимание на то, что в любом из приведенных выше определений нет указания на то, что диарея – это априори инфекционно обусловленное состояние.
Классификация диареи, используемая в клинической практике, очень удобна, что бывает достаточно редко, так как в её основе лежат два критерия: первый – клинический, по продолжительности поноса (табл. 3); второй – патогенетический, по типу поражения слизистой кишечника (табл. 4) [8, 9, 10].
В клиническом аспекте, вне зависимости от механизма и причин усиления перистальтики кишечника, острая диарея (≤14 дней) подразделяется на два вида: без примеси крови (водянистая диарея) и с примесью крови (воспалительная диарея).
Под хронической диареей, как для детей старше 12 месяцев, так и для взрослых, понимают не менее трех эпизодов дефекаций в сутки, что продолжается более одного месяца.
Острая диарея (≤14 дней), связанная с инфекционными агентами в эпидемиологическом аспекте, который не принят в нашем практическом здравоохранении, подразделяется на пять категорий: антибиотико-ассоциированный колит, диарея путешественников, диарея, обусловленная пищей или водой (food- or water-related diarrhea), диарея иммунокомпрометированных пациентов и эндемичная диарея [11]. Причем возбудители острой диареи «не привязаны» к какой-либо из указанных выше категорий и могут находиться в любой из них [12].
Этиология острой диареи (≤14 дней) в клиническом варианте без примеси крови (водянистая диарея) достаточно разнообразна (табл. 5). Следует подчеркнуть, что, невзирая на многообразие причин, острая диарея без примеси крови манифестирует в виде энтерита и чаще всего, во всяком случае, в педиатрической практике, вызывается вирусами. У детей в этиологии вирусного энтерита доминирует ротавирус, тогда как у взрослых – вирусы семейства Caliciviridae, состоящего из Sapovirus (Sapporo-like viruses [SLVs]) и Norovirus (Norwalk-like viruses [NLVs]), последний из которых является одной из причин «диареи путешественников». Это дало повод к образному названию этой инфекции – «диарея круизного корабля» [13, 14]. В структуре бактериальных инфекций при острой диарее без примеси крови в кале наиболее частыми причинами являются четыре из пяти «диареегенных штаммов» кишечной палочки [15].
Острая диарея (≤14 дней) в клиническом варианте с примесью крови (воспалительная диарея), в отличие от водянистой диареи, практически всегда обусловлена инфекционным агентом (табл. 6) и реже связана с внекишечными заболеваниями. Ключом к диагнозу инфекционного генеза острой диареи с примесью крови является наличие лихорадки [17]. Но в каждом правиле есть свои исключения. Так, при среднетяжелых и тяжелых формах неспецифического язвенного колита и терминального илеита (болезни Крона) характерно сочетание кровянистой диареи и лихорадки. Кроме того, синдром гемоколита может иметь место при некоторых системных заболеваниях соединительной ткани, например, дерматомиозите, для которых лихорадка является обязательным признаком. И, наоборот, для опаснейшей кишечной инфекции энтерогеморрагического варианта E. coli лихорадка не является атрибутивным признаком (гемоколит без лихорадки) [18]. При этом, как показал анализ драматической истории вспышки кишечной инфекции, вызванной энтерогеморрагическим вариантом Е. соli 0104:Н4, имевшей место в 2011 г. в ФРГ, применение антибактериальных химиопрепаратов потенциально несет смертельную опасность, усиливая риск возникновения гемолитико-уремического синдрома [19, 20]. В ракурсе проблемно-ориентированного подхода лечения заболеваний в детском возрасте (проф. С. П. Кривопустов, 2010) [21] вышеприведенный факт ещё раз подчеркивает, что диагноз острой кишечной инфекции не является априори показанием к назначению антибактериальной химиотерапии (табл. 6).
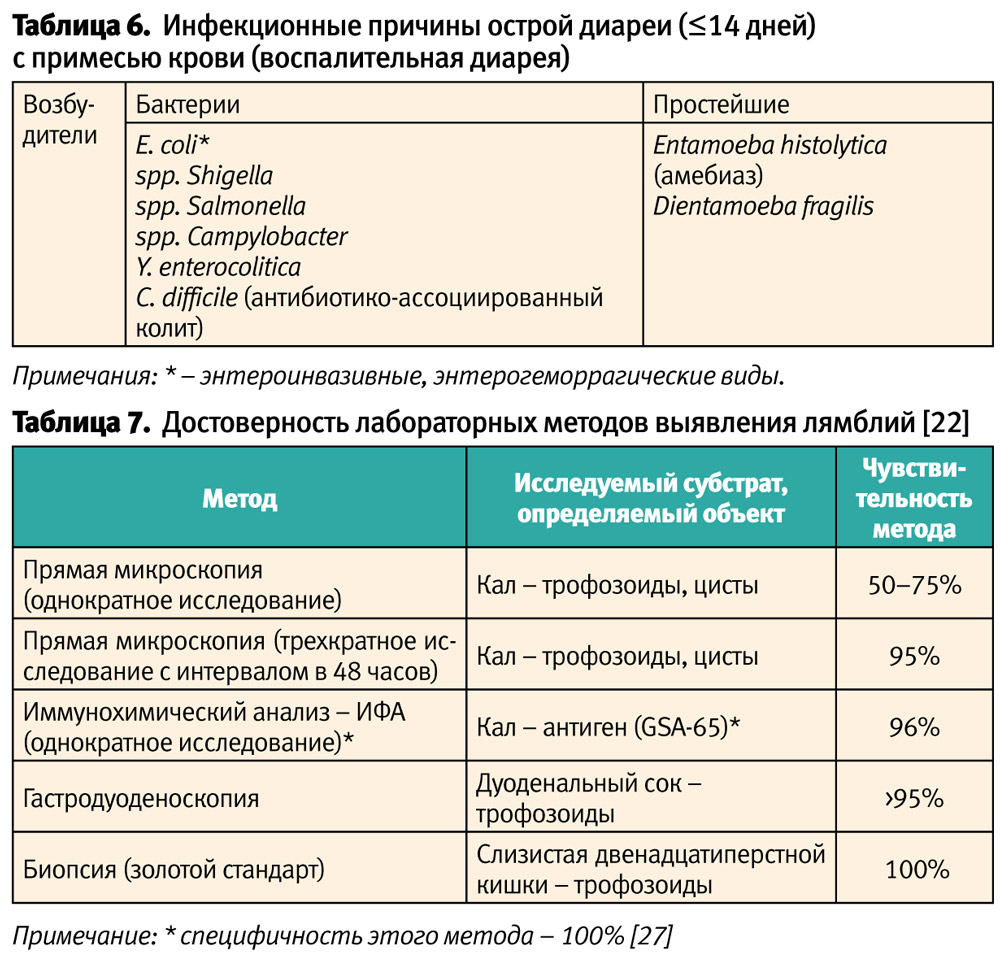
Причины острой диареи (≤14 дней):
- Практически всегда инфекционного генеза!
- Лекарственные и токсические препараты!
В случае острой диареи: копроцитограмма является первым и обязательным лабораторным исследованием, результат которого врач обязан получить в процессе «текущего рабочего дня»!
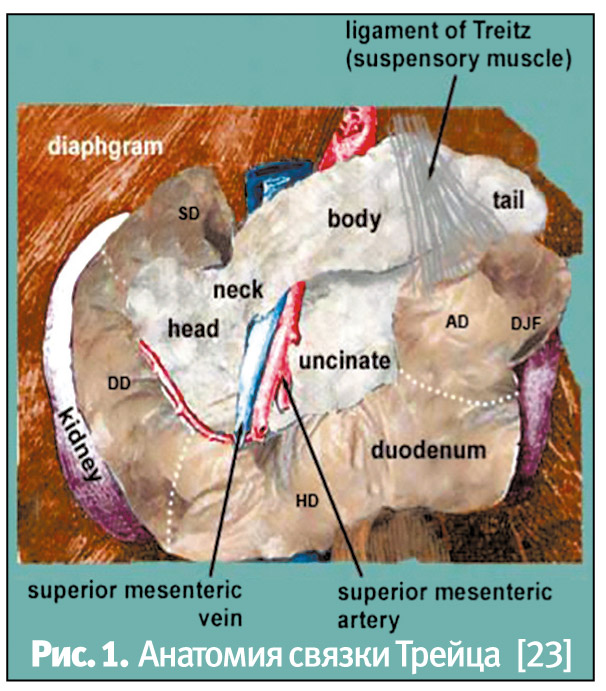
Желудочно-кишечное кровотечение также вызывает понос, что обусловлено стимулирующим влиянием крови на моторику перистальтики кишечника [17, 22]. Желудочно-кишечное кровотечение по локализации источника условно подразделяется на кровотечение из верхних и нижних отделов кишечника, где анатомической границей является связка Трейца (рис. 1, по имени чешского анатома Вацлава Трейца, 1819–1872 гг.). При оценке, является ли стул с примесью крови проявлением желудочно-кишечного кровотечения, важны следующие признаки [2, 8, 17, 22]:
- кровавая рвота, которая типична для примерно 50% пациентов при кровотечениях из верхних отделов ЖКТ;
- мелена (черный дегтеобразный стул), характерная для около 80% пациентов в случаях кровотечений из верхних отделов ЖКТ, но примерно у 1/3 пациентов источник кровотечения расположен дистальнее связки Трейца; а также в случаях, когда кровотечение длится более 14 часов, вне зависимости от локализации, может иметь место дегтеобразный стул;
- кровянистый стул (с примесью алой крови) в кале «контейнерного» типа указывает на патологию ануса (трещина, полип, сексуальное насилие); если кал жидкого типа со слизью – следует думать о язвенном колите или болезни Крона;
- стул по типу «смородинового желе» возникает при локализации источника кровотечения в дистальном отрезке подвздошной кишки или в ободочной кишке.
В любом случае, самым достоверным и практически выполнимым способом определения локализации места кровотечения является установка назогастрального зонда с последующим лаважем желудочного содержимого.
Персистирующая диарея (≥14 дней) может быть обусловлена различными заболеваниями воспалительного или наследственного происхождения. Так, персистирующую диарею способно вызывать употребление лекарственных препаратов и токсических продуктов, что ещё раз подчеркивает значимость анамнеза и знание токсикологии и фармакологии. Среди кишечных инфекций персистирующую диарею способны вызвать достаточно много инфекционных агентов, наиболее распространенными из которых являются простейшие – лямблия (Giardia lamblia), Cyclospora и амебы (Entamoeba histolytica и Dientamoeba fragilis [24], а также микробы рода иерсиний (Yersinia) и Helicobacter pylori (последний ответственен за развитие пептической язвы) [25].
Так, по данным Роспотребнадзора, среди протозоозов наиболее распространенным является лямблиоз. В 2011 г. в России зарегистрировано 77458 случаев (54,22 на 100 тыс. населения). Среди заболевших около 70% составляют дети, показатель заболеваемости детей в РФ в 2011 г. составил 203,8 на 100 тыс. детей до 17 лет. Наличие высокого риска заражения цистами лямблий подтверждается результатами санитарно-паразитологических исследований воды и смывов. Ежегодно в РФ в воде централизованного водоснабжения обнаруживают цисты лямблий. Так, в 2011 г. в «централизованной воде» были обнаружены цисты лямблий – 0,07%, в воде плавательных бассейнов – 0,02%, в смывах – 0,003% [26].
В связи с этим необходимо подчеркнуть, что для лабораторной верификации имеющейся лямблиозной инфекции рациональней применять иммунохимические методы диагностики (ИФА), позволяющие определять антиген лямблий в кале и обоснованно назначать лечение, а также объективно оценивать результаты терапии (табл. 7). Тогда как серологические методы диагностики лямблиоза, основанные на выявлении специфических иммуноглобулинов в крови, следует применять для эпидемиологических исследований, поскольку после санации организма от G. Lamblia антитела могут циркулировать на протяжении ещё 15 месяцев [27]. В случае иммунодефицита (ВИЧ, облучение, гипо- или агаммаглобулинемия) или хронических заболеваний (муковисцидоз, хронический панкреатит, ахлоргидрия) формируется носительство G. Lamblia и развитие хронической диареи.
Заболевания, сопровождающиеся возникновением хронической диареи (> 4 недель), многочисленны и, в зависимости от причин, условно разделяются на три большие группы (табл. 8).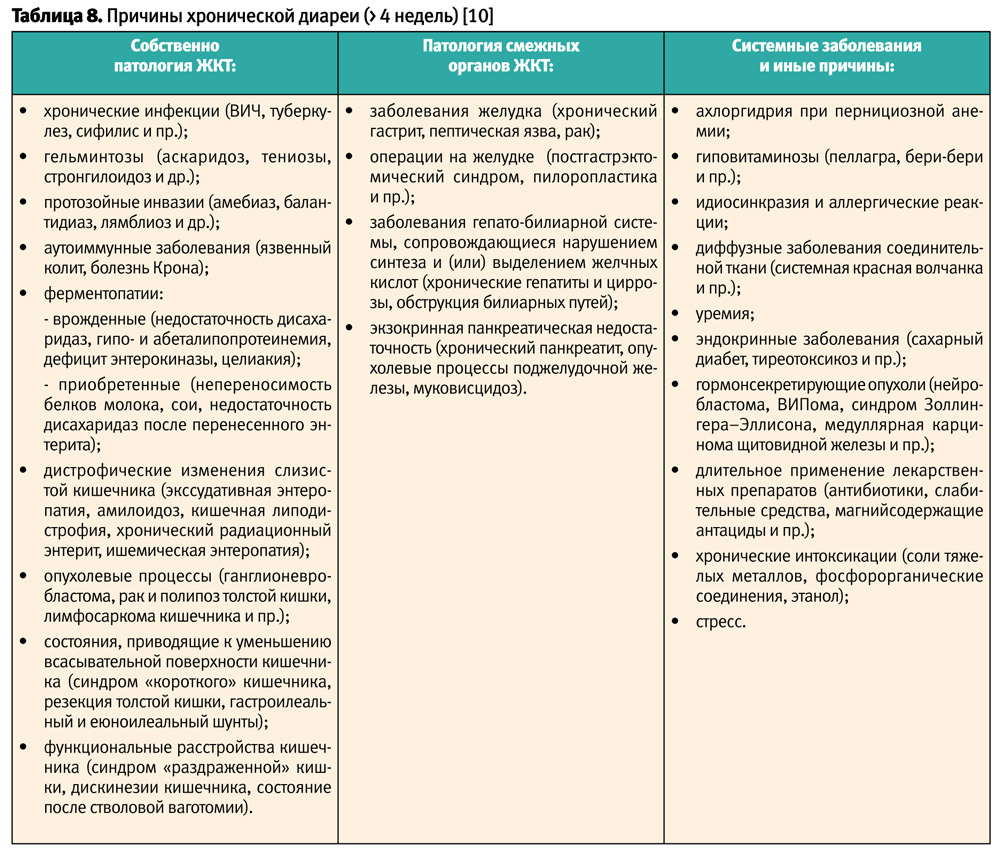
В случае хронической диареи (>4 недель) для выяснения причины, а также выбора стратегии лабораторного и инструментального обследования, прежде всего, важен сбор анамнеза и тщательный клинический осмотр пациента
Учитывая полиэтиологичность хронической диареи, универсального лабораторного теста или инструментального обследования в данной клинической ситуации нет.
Следует обратить внимание, что возникновение диареи, в том числе хронической (>4 недель) зачастую является следствием лекарственной ятрогении (антибиотики, слабительные, антациды, препараты, содержащие железо или соли магния, цитостатики и др.). Так, [цит. В. П. Булатов, А. А. Камалова] более 600 лекарств способны вызывать диарею с частотой от 1 до 83%. В связи с чем, следует кратко остановиться на одном из анахронизмов отечественной медицины – дисбактериозе [28]. Пагубность употребления понятия «дисбактериоз» и связывание его с «непонятными диареями» для врача имеет несколько аспектов. Первый – это «конфликт интересов», когда врач сознательно или «по наитию» скрывает внутрибольничную кишечную инфекцию или ятрогению. Второй – задержка времени установления истинного диагноза (причины диареи). Третий – трата материальных и экономических ресурсов на бессмысленные с точки зрения диагностической значимости микробиологические исследования.
Заключение
Диарея сопровождает множество заболеваний и не является априори клиническим признаком кишечной инфекции. В диагностике диарей наиболее ценным инструментом в арсенале врача является тщательно собранный анамнез и клинический осмотр больного, позволяющие значительно сузить круг предполагаемых заболеваний и составить план рационального лабораторного и инструментального обследования. Остро возникшая диарея требует исключения инфекционной этиологии заболевания и обязательного копроцитологического обследования, тогда как хроническая диарея чаще всего имеет неинфекционную природу.
Список литературы находится в редакции
Детальніше
НЕОНАТАЛЬНЫЕ ГЕПАТИТЫ
 Гепатит (ἡπατῖτις от греч. ἥπαρ – печень) – воспалительное заболевание печени различной этиологии. Гепатит, симптомы которого развились у ребенка до 2–3-месячного возраста (чаще на первом месяце), считается неонатальным или перинатальным
Гепатит (ἡπατῖτις от греч. ἥπαρ – печень) – воспалительное заболевание печени различной этиологии. Гепатит, симптомы которого развились у ребенка до 2–3-месячного возраста (чаще на первом месяце), считается неонатальным или перинатальным
Гепатит – заболевание инфекционной природы. Причинами возникновения гепатита у новорожденных могут быть вирусы (вирус простого герпеса – ВПГ, цитомегаловирус – ЦМВ, Varicella–Zoster-вирус, другие герпес-вирусы, вирус гепатита В (ВГВ), вирус краснухи, ECHO- и Коксаки-вирусы и др.), Treponema pallidum, бактерии (микобактерии туберкулеза, Listeria monocytogenes, кишечная палочка, стафилококк и др. бактерии-возбудители сепсиса), Toxoplasma gondii. Следует отметить, что передача вируса гепатита С от матери ребенку происходит достаточно редко, а гепатит при этом развивается не в первые месяцы жизни. Риск развития вирусного гепатита А у новорожденных также очень низкий. Вирус иммунодефицита человека (ВИЧ), который передается от матери ребенку, не вызывает гепатит. Однако у детей, рожденных ВИЧ-инфицированными матерями, значительно выше риск развития неонатального гепатита, чем в популяции, что объясняется иммунодефицитом у матери, наличием у нее оппортунистических и других инфекций, многие из которых также вызывают перинатальные инфекции (ВПГ, ЦМВ, ВГВ, сифилис, токсоплазмоз и др.), сопровождающиеся гепатитом.
Возбудитель попадает к плоду внутриутробно или во время родов от матери, у которой есть острый или хронический (персистирующий) инфекционный процесс, или латентное (бессимптомное) носительство микроорганизмов. Наиболее опасным для плода является первичное инфицирование матери во время беременности. Вторичная инфекция (реинфекция или реактивация хронической инфекции во время беременности) также может привести к инфицированию плода, но при этом риск перинатальной передачи возбудителя ниже, чем при первичном инфицировании. Медицинские манипуляции, ятрогенные вмешательства, патология беременности, нарушающие целостность плаценты, плодных оболочек, кожи плода также способствуют передаче возбудителя.
Пути инфицирования плода: гематогенный (диаплацентарный); через околоплодные воды – восходящий (из влагалища) или нисходящий (из маточных труб); трансмембранный (через плодные оболочки); контактный (интранатальный) – при контакте кожи и слизистых оболочек плода с родовыми путями, заглатывании инфицированного содержимого родовых путей.
Антенатальное инфицирование гематогенным путем возможно вирусами, простейшими паразитами и некоторыми бактериями (листерии, микобактерии). Риск гематогенного заражения плода во многом зависит от барьерной функции плаценты. При плацентарной недостаточности у матери с очагами хронической инфекции или любым острым инфекционным процессом резко увеличивается возможность развития генерализованной инфекции плода. Экстрагенитальные заболевания матери и осложнения беременности (гестозы), увеличивающие проницаемость плаценты, значительно повышают риск развития инфекций у плода. С другой стороны, возбудители антенатальных инфекций могут непосредственно поражать плаценту, вызывая плацентит, что, в свою очередь, приводит к плацентарной недостаточности и прерыванию беременности, инфицированию плода.
Инфекции мочеполовой системы матери (пиелонефрит, сальпингит, сальпингоофориты, эндоцервициты, эндометриты, эрозии шейки матки, вульвовагиниты, бактериальный вагиноз и т. д.) вызываются стафилококками, стрептококками, диплококками, грам-отрицательной кишечной флорой, листериями, бруцеллами, микобактериями туберкулеза. В половых путях они могут паразитировать и вызывать хронические воспалительные процессы ЦМВ, ВПГ (чаще II типа). Инфицирование плода этими возбудителями может происходить через околоплодные воды восходящим и нисходящим путем, а также контактным путем при прохождении плода по родовым путям.
Внутриутробные инфекции могут быть вызваны как одним возбудителем, так и несколькими возбудителями одновременно (часто оказывается вирусно-бактериальная ассоциация); микст-инфекция значительно увеличивает тяжесть течения заболевания и ухудшает прогноз.
Постнатальное инфицирование такими возбудителями как ЦМВ, ВПГ и другими вирусами, может происходить как от матери, так и от других лиц, ухаживающих за ребенком. Передача некоторых возбудителей неонатального гепатита также возможна через препараты крови.
Для уточнения этиологии неонатального гепатита нужно анализировать результаты обследования матери и ребенка
Степень поражения ткани печени при гепатите различными возбудителями варьирует от умеренных диффузных воспалительных изменений (при ЦМВ, ВГВ) до фокального или массивного гепатонекроза (при ВПГ). Возможно также развитие абсцессов печени при пупочном сепсисе, чаще колибациллярной или стафилококковой этиологии.
Клинические проявления гепатита выявляются чаще в первые недели жизни. Для гепатита характерны: гепатомегалия, желтуха, темная моча, обесцвеченный кал, явления кровоточивости, неспецифические симптомы инфекционного токсикоза. Желтуха, как правило, возникает после первых суток жизни. Цвет кожи варьирует от бледного с желтовато-зеленоватым оттенком до интенсивно желтого, чаще с зеленоватым оттенком. При некоторых инфекциях (например, ЦМВ-инфекции, ВПГ-инфекции, листериозе и др.) на коже может выявляться различная сыпь – макулы, папулы, везикулы. Как результат гипопротеинемии, могут возникать отеки. К общим симптомам инфекционного токсикоза относят: снижение аппетита или отказ от еды, угнетение рефлексов периода новорожденности, вялость, метеоризм, рвота. Гепатомегалия (печень выступает из-под края реберной дуги более, чем на 3 см) может сопровождаться спленомегалией. Геморрагический синдром, полиорганная недостаточность, тяжелые метаболические нарушения являются непосредственными причинами смерти ребенка с неонатальным гепатитом.
Лабораторные признаки поражения печени:
- гипербилирубинемия за счет прямой и непрямой фракции – признак нарушения выделительной функции печени;
- повышение уровня трансаминаз (более специфична аланинаминотрансфераза, менее специфична аспартатаминотрансфераза) – признак цитолиза;
- повышение уровня щелочной фосфатазы и гамма-глутамилтранспептидазы – признак холестаза;
- гипокоагуляция и гипоальбуминемия – признак нарушения белково-синтетической функции печени.
Для уточнения этиологии неонатального гепатита нужно анализировать результаты обследования матери и ребенка. Выявлять возбудитель можно прямыми методами в крови, моче, везикулах, смывах влагалища микробиологическими или вирусологическими методами на средах или культурах клеток. К прямым методам также относится полимеразная цепная реакция (ПЦР), которая позволяет выявить наличие генетического материала (ДНК или РНК) возбудителя, содержащегося даже в небольшом количестве в биологических жидкостях. Хотя чувствительность метода ПЦР очень велика, трактовать полученные результаты желательно вместе с анализом титра специфических антител.
Серологические (иммунологические) методы позволяют выявлять антитела к возбудителям следующими тестами: иммуноферментный анализ (ИФА), реакции прямой и непрямой иммунофлюоресценции, реакция связывания комплемента, реакция агглютинации, реакция торможения гемагглютинации и другие.
С помощью метода ИФА возможно качественно и количественно выявлять иммуноглобулины (Ig) классов M, G, A к отдельным возбудителям. Выявление специфических IgM указывает на наличие острого заболевания у человека. Учитывая, что IgG могут проникать через плаценту от матери к плоду, наличие у новорожденного ребенка специфичных для возбудителя IgG не подтверждает внутриутробное инфицирование ребенка. Элиминация материнских специфических антител из крови ребенка может длиться от нескольких недель до 18 месяцев. Если у ребенка в динамике значительно увеличивается титр специфических IgG, то это свидетельствует в пользу инфекции.
Определение авидности антител помогает различить IgG матери и ребенка. В первые 1–1,5 месяца после инфицирования у человека появляются антитела низкоавидные, то есть демонстрирующие низкую степень сродства с антигеном. В ходе естественного развития иммунного ответа в организме человека проявляются высокоавидные антитела. При вторичном инфицировании за счет этих антител развивается быстрый вторичный иммунный ответ. Выявление у матери или у ребенка IgМ и/или IgG с низкой авидностью свидетельствует о первичном недавнем инфицировании. Если у матери в крови одновременно имеются и IgМ, и высокоавидные IgG, это указывает на вторичную инфекцию, или обострение (реактивацию) латентной инфекции. Наличие у ребенка высокоавидных IgG указывает на то, что это материнские антитела.
Биопсия печени с последующим исследованием биоптата морфологическим и вирусологическими методами на практике используется крайне редко, хотя является достаточно важным дифференциально-диагностическим тестом для исключения внутрипеченочной атрезии желчных ходов и определения степени фиброза.
Клинико-лабораторные признаки, характерные для неонатального гепатита инфекционной природы, также могут наблюдаться при токсическом действии лекарственных веществ на печень, наследственных заболеваниях и билиарной атрезии.
Дифференциальную диагностику неонатального гепатита проводят с наследственными нарушениями обмена веществ (галактоземия, тирозинемия), наследственными заболеваниями (дефицит a1-антитрипсина, муковисцидоз, неонатальный гемохроматоз, болезнь Байлера) и синдромами (Алажилля, Цельвегера), для которых также характерны гепатомегалия, гипербилирубинемия, повышение трансаминаз, нарушение функции печени. Дифференциальная диагностика неонатального гепатита инфекционной природы с наследственными заболеваниями очень важна, так как определяет правильную тактику ведения и прогноз.
Следует дифференцировать гепатит новорожденных и атрезию желчевыводящих путей (билиарную атрезию) – врожденную патологию, при которой желчевыводящие пути непроходимы или отсутствуют. При этом следует понимать, что билиарная атрезия может быть результатом гепатита плода, перенесенного во II триместре беременности. Реконструктивная хирургическая коррекция с созданием протоков (портоэнтеростомия, процедура Касаи) или пересадка печени являются способами лечения билиарной атрезии. Реконструктивную операцию желательно производить не позже, чем в двухмесячном возрасте, поскольку откладывание операции приведет к развитию цирроза, что резко снижает выживаемость детей. Даже при хирургическом вмешательстве вероятность смертельного исхода при билиарной атрезии – более 50%.
Хотя чувствительность метода ПЦР очень велика, трактовать полученные результаты желательно вместе с анализом титра специфических антител
Идиопатический неонатальный гигантоклеточный гепатит – воспалительное поражение печени, этиология которого не установлена, и при этом исключены известные возбудители гепатита новорожденных, наследственные заболевания и нарушения обмена веществ, билиарная атрезия.
Лечение неонатального гепатита включает этиотропную терапию, если выявлены ВПГ или Varicella–Zoster-вирус, Treponema pallidum, бактерии (микобактерии туберкулеза, Listeria monocytogenes, или возбудители сепсиса), Toxoplasma gondii. Синдромное лечение включает: обеспечение достаточной калорийности энтерального или парентерального питания (120–150 ккал/кг); поддержание водно-электролитного баланса; урсодезоксихолиевую кислоту (желчная кислота, способствующая снижению содержание холестерина в желчи, оказывает влияние на энтерогепатическую циркуляцию желчных солей, уменьшая реабсорбцию в кишечнике эндогенных токсичных соединений) в дозе 10–15 мг/кг в сутки; колестирамин (препятствует всасыванию желчных кислот и холестерина в кишечнике, связывая их с образованием комплексов, выводящихся с каловыми массами, повышает синтез желчных кислот из холестерина в печени, снижает концентрацию липидов низкой плотности, холестерина и желчных кислот в плазме, уменьшает кожный зуд) в дозе 0,25–0,5 г/кг в сутки в 3 приема; введение жирорастворимых витаминов (A, E, K, D). Назначение глюкокортикоидов противопоказано.
Прогноз неонатального инфекционного гепатита: у 35% детей развивается цирроз печени, 10–15% детей погибают в первые месяцы жизни и около 20% умирают в возрасте до 24 месяцев после рождения.
Литература
- Аряев Н. Л. Неонатология: учебник / Н. Л. Аряев. – Одесса.: Одес. гос. мед. ун-т, 2006. – 836 с.
- Аряєв М. Л. Вроджений вірусний гепатит (P35.3) / М. Л. Аряєв, Н. В. Котова// Неонатологія, хірургія та перинатальна медицина. – 2011. – № 2. – С. 121-126.
- Вірусний гепатит В у новонароджених: навч. метод. посібник / За ред. Гиріна В. М. [І. В. Дзюблик, Є. Є. Шунько, О. Т. Лакша, Г. І. Гречень] // – К.: – 2001. – 85 с.
- Congenital Infections, Part I: Cytomegalovirus, Toxoplasma, Rubella, and Herpes Simplex / C. Tian, S. Asad Ali, J. H. Weitkamp // NeoReviews. – 2010. – N 11 (8). – P. e436-e446.
- Neonatology / ed. by R. A. Polin, J. M. Lorenz. – Cambridge University Press, 2008. – 585 p.
- Update on TORCH Infections in the Newborn Infant: Advances in Congenital Infections // – Режим доступу: http://www.medscape.com/viewarticle/472409_7 – Назва з екрану.
- Viral Infections and Pregnancy / [T. Marino, B. Laartz, S. E Smith, et al.]// – Мedscape, 2011 – Режим доступу: http://emedicine.medscape.com/article/235213-overview – Назва з екрану.

СРАВНИТЕЛЬНАЯ ОЦЕНКА ЭФФЕКТИВНОСТИ ПРИМЕНЕНИЯ ПРОБИОТИКА ЭНТЕРОЖЕРМИНА В ЛЕЧЕНИИ МЛАДЕНЧЕСКОЙ КИШЕЧНОЙ КОЛИКИ У ДЕТЕЙ
 Частота возникновения младенческой кишечной колики (МКК) у детей грудного возраста составляет от 10–15% до 30–70% [1, 4]. МКК возникает на фоне полного благополучия и удовлетворительного питания и проявляется приступами раздражительности, беспокойства или плача в общей сумме в течении 3 часов в день и возникающих не менее 3 раз в неделю на протяжении 3 недель («правило трех») [5]
Частота возникновения младенческой кишечной колики (МКК) у детей грудного возраста составляет от 10–15% до 30–70% [1, 4]. МКК возникает на фоне полного благополучия и удовлетворительного питания и проявляется приступами раздражительности, беспокойства или плача в общей сумме в течении 3 часов в день и возникающих не менее 3 раз в неделю на протяжении 3 недель («правило трех») [5]
Этиопатогенез младенческой кишечной колики до конца не выяснен. Среди основных причин называют: морфофункциональную незрелость периферической иннервации кишечника, дисфункцию центральной регуляции, позднее созревание ферментативной системы органов ЖКТ, повышенное газообразование, синдром мальабсорбции, нарушения в формировании микробиоценоза кишечника, характер питания матери, аллергические и псевдоаллергические реакции, переход с естественного вскармливания на искусственное, включение в рацион пищевых добавок, биопсихосоциальные обстоятельства [1–3, 6].
Интестинальной флоре придается большое значение в формировании иммунной системы ЖКТ. Микробная стимуляция на первом месяце жизни модифицирует иммунный ответ, приводя к развитию устойчивости ребенка к окружающим аллергенам. Соответственно, при нарушении формирования микробиоценоза кишечного тракта, при появления в нем условно-патогенных микроорганизмов могут происходить изменения, приводящие к нарушению толерантности и, как следствие, к возникновению аллергических заболеваний. Учитывая вышеизложенное, была поставлена задача оценить эффективность применения пробиотиков в лечении младенческой кишечной колики.
Целью исследования стала оценка эффективности препарата Энтерожермина (Sanofi Aventis) у детей с младенческой кишечной коликой. Препарат представляющий собой суспензию спор Bacillus clausii, которые являются естественными сапрофитами и ингибируют развитие патогенных микроорганизмов путем конкурентного связывания с рецепторами слизистой оболочки кишечника. Благодаря высокой резистентности к химическим и физическим агентам споры Bacillus clausii неповрежденными проходят через барьер желудочного сока и желчных кислот в кишечник, где они трансформируются в метаболически активные вегетативные формы [7].
Проведено проспективное рандомизируемое контролируемое исследование, в котором принимали участие 60 детей в возрасте от 28 до 90 дней, наблюдавшихся в детской поликлинике на базе областной детской клинической больницы г. Одессы. Родители включенных в исследование детей дали информированное согласие на участие в исследовании.
Критерии включения: дети с наличием младенческой кишечной колики; возраст: 28–90 дней; пол: 28 мальчиков и 32 девочки; информированное согласие родителей на участие пациента в исследовании. Критерии исключения: нахождение ребенка на искусственном вскармливании; отсутствие согласия на участие в исследовании; наличие синдрома кишечной колики на фоне органической патологии ЖКТ; острые заболевания других органов и систем; наличие в анамнезе факта применения антибиотика или другого пробиотика за последние 10 дней; отказ выполнять основные процедуры исследования и выполнять режим приема препарата.
Диагноз основного заболевания верифицировался на основании анализа клинико-анамнестических данных, результатов объективного обследования, а также с использованием инструментальных и лабораторных методов для исключения органической природы заболевания.
Возраст детей находился в пределах от 28 до 90 дней (средний возраст 56,2±20 дней), среди исследуемых было 32 девочки и 28 мальчиков. По результатам рандомизации пациенты были разделены на 2 группы, 30 детей основной группы получали в качестве терапии МКК препарат Энтерожермина (sanofi-aventis) в дозировке 1 флакон 1 раз в сутки, который разводился в воде или молоке, длительность приема препарата составила 28 день.
В группу сравнения входили 30 детей, которые в качестве лечения получали пеногасители. Статистически значимых клинических, возрастных и половых различий между группами не было (p>0,05). Длительность наблюдения составила 28 дней.
Эффективность лечебного действия препарата оценивали по суммарной суточной длительности эпизодов колики, их количеству возникновения за день, а также по субъективной интегральной шкале оценки удовлетворенности результатами лечения. (Integrative Medicine Patient Satisfaction Scale, IMPSS). IMPSS широко распространена в исследованиях и описывает удовлетворенность пациента (или родителей пациента) лечением. Она состоит из 5 пунктов: всецело удовлетворен, удовлетворен; отношусь нейтрально, не удовлетворен, крайне не удовлетворен. В первый день проводилось общеклиническое обследование ребенка, сбор жалоб, анамнеза болезни и жизни и рандомизация пациентов.
Эффективность проводимой терапии оценивалась на 2, 7, 14, 21 и 28 день от начала лечения. Безопасность и переносимость препарата оценивали по наличию или отсутствию ожидаемых побочных действий, включая аллергические реакции и случаи индивидуальной непереносимости.
Статистическая обработка полученных данных выполнена с использованием стандартных пакетов программ Microsoft Excel.
Установлено, что на 14 день в основной группе (дети, получавшие препарат Энтерожермина) отмечается большее уменьшение длительности (93,7±32,6 мин/день) и частоты колик (6,25±2,18 эпизодов в день), чем в группе сравнения (159,5±34,07 мин/день и 10,6±2,28 эпизодов в день соответственно, p<0,001). В конце лечения средняя продолжительность кишечных колик в основной группе и группе сравнения составила соответственно 52,8±31 мин/день и 148,1±35,8 мин/день. В последний день наблюдения средняя частота возникновения кишечной колики в основной группе составила 3,5±2 эпизодов в сутки, тогда как в группе сравнения это показатель был достоверно выше – 9,9±2,4 эпизодов в сутки (p<0,001).
У 28 детей (93,3 %) основной группы среднесуточная продолжительность приступов колик уменьшилась к концу наблюдения более чем в 2 раза, тогда как в группе сравнения только у 3 детей (10%) наблюдалось сокращение длительности приступов.
Таким образом, применение препарата Энтерожермина, по сравнению с симетиконом, приводит к более быстрому и выраженному уменьшению длительности и частоты эпизодов младенческой кишечной колики у грудных детей.
Субъективную оценку «всецело удовлетворен» в основной группе поставило 26 человек (родители пациентов), оценку «удовлетворен» – 4. В группе сравнения оценку «всецело удовлетворен» поставило 18 человек (матерей), оценку «удовлетворен» – 12.
В ходе мониторинга нежелательных явлений побочное действие препарата в виде аллергических реакций, случаев индивидуальной непереносимости зафиксировано не было. Препарат Энтерожермина хорошо переносился детьми, отказов детей от приема препарата не было.
Выводы:
Приём препарата Энтерожермина (Sanofi-aventis) способствует положительной динамике клинических проявлений и суточной длительности младенческой кишечной колики у детей.
Препарат Энтерожермина отличается хорошей индивидуальной переносимостью, низкой частотой развития побочных реакций и не препятствует формированию приверженности терапии.
Литература
- Lucassen P. L. B., Assendelft W. J. J., van Eijk J. T. M., Gubbels J. W., Douwes A. C., van Geldrop W. J. Systematic review of the occurrence of infantile colic in the community // Arch. Dis. Child. – 2001. – V. 4. – P.: 398-403.
- Low Plasma Cholecystokinin Levels in Colicky Infants / Huhtala Virpi, Lehtonen Liisa, Uvnäs-Moberg Kerstin, Korvenranta Heikki // Journal of Pediatric Gastroenterology and Nutrition. – 2003. – V. 37, N 1. – P.: 42-46
- Sondergaard C., Henriksen T.B., Obel C., Wisborg K. Smoking during pregnancy and infantile colic // Pediatrics. – 2001. – V. 108, N 2. – P: 342-348.
- Килгур Т., Уэйд С. Колики у детей грудного возраста // Доказательная медицина. – 2005. – № 4. – С. 629-661.
- Wessel M. A., Cobb J. C., Jackson E. B., Harris G. S., Detwiler A. C. Paroxismal fussing in infancy, sometimes called «colic» Pediatrics. 1954;14:421-435
- Leung A. K., Lemau J. F. Infantile colic: a review // J. R. Soc. Health. – 2004. – Vol. 124, № 4. – P. 162.
7. Арцезе А. Пробиотическая активность Bacillus clausii при диарее у детей // Современная педиатрия. – 2008. – 4(21). – С. 166-169.
Детальніше