
ПРЕДИКТИВНАЯ МЕДИЦИНА В ГЕМАТОЛОГИИ
В области современной клинической гематологии идет интенсивный поиск маркеров предрасположенности, ранней диагностики и прогноза течения гематологических и, в первую очередь, онкогематологических заболеваний. В основе развития злокачественных заболеваний системы крови лежит наследственная предрасположенность, которая ассоциируется с генетическими факторами организма. Именно генетически детерминированные характеристики являются устойчивыми и представляют наибольшую прогностическую ценность в клиническом аспекте. Раскрытие механизмов лейкемогенеза и роли генетических факторов в этом процессе является одной из приоритетных задач современной гематологии. Существуют различные схемы лейкемогенеза, отражающие последовательность молекулярных событий, приводящих к возникновению опухоли. Для острых лейкемий предложена модель «генетического груза»: развитию опухоли предшествует накопление в геноме мутаций, повышающих пролиферативный потенциал клетки и/или блокирующих механизмы апоптоза. Этот «генетический груз» может неопределенно долгое время компенсироваться функционированием системы генов-супрессоров опухолевого роста. Последующие мутации в генах-супрессорах или делеции интактных аллелей протоонкогенов приводят к интенсивной пролиферации бластных клеток. Кроме того, накапливающиеся в геноме нарушения могут повлечь за собой качественные изменения в молекулярных механизмах регуляции основных процессов, происходящих в клетке, что также может привести к индукции опухолевого процесса. Малигнизация нормальной клетки является результатом каскадного накопления определенных изменений в ее генетическом материале. Нарушения в структуре генов могут происходить без видимых воздействий. Каждую секунду в организме человека происходит деление около 25 млн. клеток. Этот процесс находится под строгим контролем, осуществляемым комплексом молекулярных систем в определенных органах и в точное время. Все возникающие нарушения фиксируются и устраняются системами репарации клеточного генома. Однако в некоторых случаях восстановление нормальной структуры измененного гена может не состояться, кодируемый белковый продукт и его свойства изменяются. Если эта аномалия имеет принципиальный характер и затрагивает ключевые гены (потенциальные онкогены), то становится возможной трансформация клетки.
К генетическим детерминантам клетки, вовлеченным в канцерогенез, относятся протоонкогены – нормальные клеточные гены, участвующие в ключевых процессах жизнедеятельности клетки, регуляции ее транскрипции, роста, клеточного цикла, в передаче сигнала и т. д. В случае структурных изменений или при повышении уровня экспрессии протоонкогенов нарушается контроль нормального клеточного роста и дифференцировки, что приводит к трансформации клетки. Существуют два основных механизма нарушения функции протоонкогенов в лейкемических клетках:
- аберрации, приводящие к структурным изменениям протоонкогена и формированию гибридных (химерных) генов. В результате таких аберраций происходят качественные изменения белков, приобретающих онкогенную активность;
- генетические перестройки, сопровождающиеся переносом протоонкогена в область генов иммуноглобулина (Ig) или генов рецептора Т-лимфоцитов (TCR). Данные аберрации, характерные для зрелой В- и Т-клеточной острой лимфобластной лейкемии, приводят к состыковке кодирующих последовательностей протоонкогена с сильными промоторами генов TCR или Ig, следствием чего является повышение уровня экспрессии протоонкогена.
В настоящее время известно более 200 различных протоонкогенов, изменения структуры или гиперэкспрессия которых приводят к продукции дефектных онкобелков. Это, в свою очередь, нарушает взаимосвязь белков-партнеров, что, в конечном счете, ведет к трансформации клетки.
Таким образом, каждая нормальная клетка содержит набор протоонкогенов, необходимых для нормальной жизнедеятельности клетки и выполняющих множество разнообразных функций. Под воздействием определенных факторов изменяется генетическая программа клеток, и они начинают бесконтрольно делиться; при этом в образовавшейся опухоли удается выявить определенные изменения либо в структуре одного (или нескольких) протоонкогенов, либо в изменении их локализации, либо в увеличении числа копий (амплификации), либо в увеличении экспрессии, либо в появлении мутаций.

В геноме человека протоонкогены локализованы практически на всех хромосомах. Многие из них располагаются в участках хромосом, претерпевающих неслучайные изменения при злокачественных новообразованиях. Оказалось, что ранние перестройки хромосом являются важным условием для активации латентных онкогенов генома при их транспозиции в новое генетическое окружение. Так, наблюдавшиеся при лейкемиях нарушения в хромосомах 6, 8, 9, 15, 17, 20, 22 интересны с той точки зрения, что именно в этих хромосомах локализуются клеточные протоонкогены, которым отводится большая роль в процессах малигнизации. При транслокации могут происходить изменения положения клеточных онкогенов с последующим повышением их экспрессии. Отмеченные в ряде случаев моносомии или трисомии этих хромосом также могут играть роль в малигнизации в результате изменения соотношения нормальных и мутировавших онкогенов в клетке. Возможно, что при трисомиях появляются дополнительные копии генов и увеличивается доза активированного онкогена – онкоген многократно реплицируется, и в клетке оказывается увеличенным число его копий, что ведет как к малигнизации, так и к прогрессии.
К генетическим детерминантам клетки относятся также гены-супрессоры (антионкогены) – гены, кодирующие ключевые регуляторные белки, потеря которых влечет за собой нарушения контроля пролиферации. Наряду с активацией онкогенов, нарушения работы генов-супрессоров опухоли являются решающими в инициации туморогенных процессов, влияя на прохождение клеточного цикла, регулируя дифференцировку и программированную гибель клеток (апоптоз).
Структурные и функциональные изменения в онкосупрессорах, как и в онкогенах, могут быть следствием точечных мутаций в кодирующих и регуляторных областях гена, вставок или делеций, вызывающих нарушения процесса считывания белков, изменение их конфигурации или модуляцию белковой экспрессии.
С расшифровкой генома в 2006 г. и завершением программы «геном человека» интерес гематологов к изучению генетических механизмов развития гематологической патологии значительно возрос. Использование в клинической практике современных высокотехнологичных молекулярно-генетических методов позволило расшифровать генетические механизмы лейкемогенеза, этиопатогенеза различных заболеваний системы крови и кроветворных органов. Спектр гематологических заболеваний широк и включает в себя анемии (талассемии, серповидно-клеточная анемия); геморрагические диатезы, включающие коагулопатии (гемофилия, болезнь Виллебранда), нарушения гемостаза (геморрагический васкулит), тромбоцитопении, тромбоцитопатии и гемобластозы (хроническая лимфоцитарная лейкемия).
Большинство гематологических заболеваний являются моногенными, поэтому генетическое тестирование является важным не только для медико-генетического консультирования, но и для диагностики, дифференциальной диагностики гематологических заболеваний, а также их лечения.
Гемофилия
Одним из наиболее известных моногенных гематологических заболеваний является гемофилия, часто называемая болезнью королей. Гемофилия A является наследственным заболеванием крови и характеризуется дефицитом белка свертывания крови, известного как Фактор VIII, который приводит к аномальному кровотечению. Вызывается данное заболевание мутацией гена HEMA на X-хромосоме. Ген HEMA кодирует фактор VIII, который синтезируется, главным образом, в печени и является одним из многих факторов, вовлеченных в процесс свертывания крови. В настоящее время широко дискутируется вопрос о целесообразности применения генной терапии в лечении гемофилии.
Серповидно-клеточная анемия (СКА)
Это гемоглобинопатия, которая характеризуется эпизодическими болями, хронической гемолитической анемией и тяжелыми инфекциями, обычно начинающимися в детстве. СКА наследуется по аутосомно-рецессивному типу и вызывается точечной мутацией в гене гемоглобина бета (НВВ). Эта мутация приводит к продукции гемоглобина со структурными дефектами – HbS. Гемоглобин является протеином, переносящим кислород. При определенных условиях, например, при низком содержании кислорода или высоком содержании гемоглобина, у гомозиготных носителей HbS эритроциты приобретают серповидную форму, что приводит к блокировке небольших кровяных сосудов, развитию болевого синдрома и травмированию внутренних органов.
Талассемия
Заболевание вызывается наследственным дефектным синтезом гемоглобина, количественной гемоглобинопатией. Зрелый гемоглобин состоит из двух a- и двух b-полипептидных цепей. Существуют две копии гена a-гемоглобина (HBA1 и HBA2), каждая их которых кодирует a-цепь. Ген бета-гемоглобина (HBB) кодирует b-цепь. При a-талассемии в результате мутаций в генах HBA1 и HBA2 нарушается синтез a-цепи, избыток b-цепи недостаточно связывает кислород, приводя к низкой концентрации его в тканях (гипоксемия). Аналогично при b-талассемии нарушается синтез b-цепей. Однако, избыток a-цепи может привести к формированию нерастворимых частиц в эритроцитах. Эти частицы приводят к гибели эритроцитов и их предшественников, вызывая развитие тяжелой формы анемии. В связи с интенсивным удалением в селезенке поврежденных эритроцитов, развивается спленомегалия. Делеции в генах HBA1 и/или HBA2 являются причиной большинства случаев a-талассемии и определяют тяжесть клинических проявлений заболевания. Потеря одного или двух генов обычно является бессимптомной, в то время как потеря всех четырех генов является фатальной для будущего ребенка. В гене HBB насчитывается более 100 мутаций, однако делеции при этом редки. Тяжесть клинических проявлений заболевания зависит от локализации мутации; так, сплайсинг-мутации и мутации в области промотора HBB гена приводят к сокращению, а не полному отсутствию цепей b-глобина и, как результат, – к более благоприятному течению заболевания. Нонсенс-мутации и мутации со сдвигом рамки считывания приводят к отсутствию продукции цепей b-глобина, что обусловливает тяжелое течение болезни. В настоящее время оценивается потенциал генотерапии для лечения данного заболевания.
Гемоглобинопатия С
Гемоглобинопатия С характеризуется нарушением синтеза нормального Нb с образованием аномального глобина (НbС) вследствие мутации в гене НВВ. Наследуется по аутосомно-рецессивному типу. НbС имеет в своем составе аномальный глобин, что понижает кислородосвязывающую функцию Нb и нарушает пластичность эритроцитов, в результате у гомозигот формируются серповидноклеточные эритроциты.
Гемоглобинопатия E
Гемоглобинопатия Е характеризуется нарушением синтеза нормального Нb с образованием аномального глобина (НbЕ) вследствие мутации G79A в гене НВВ. Наследуется по аутосомно-рецессивному типу. Мутация вызывает аномальный сплайсинг некоторых молекул транскрипта, что препятствует трансляции полученной мРНК. Это приводит к выраженному дефициту бета-цепей глобина и напоминает бета-талассемию. При гетерозиготной форме гемоглобинопатии Е наблюдаются легкая микроцитарная гипохромная анемия и наличие мишеневидных эритроцитов. Гомозиготная форма также протекает сравнительно легко, с резко выраженным микроцитозом, обычно без спленомегалии. У смешанных гетерозигот (HbE-, tnf-талассемия) болезнь протекает тяжелее, наблюдается спленомегалия.
Анемия Фанкони
Заболевание наследуется аутосомно-рецессивно и проявляется анемией, лейкоцитопенией и тромбоцитопенией, а также ломкостью хромосом и повышенной чувствительностью к химическим мутагенам. Характерны стигмы дизэмбриогенеза и предрасположенность к развитию злокачественных новообразований. Исследования выявили несколько различных генов (FANCB, FANCC, BRCA2, FANCD2, FANCE, FANCF, XRCC9, FANCI, BRIP1, PHF9, FANCM, PALB2, RAD51C и SLX4), мутации которых обуславливают анемию Фанкони. Один из генетических вариантов заболевания – тип С – обусловлен мутацией гена FANCC, участвующего в ответе клетки на повреждение ДНК.
Болезнь фон Виллебранда
Это нарушение свертывания крови вследствие недостаточной активности фактора фон Виллебранда – гликопротеида, который находится в плазме в виде смеси олигомеров и обеспечивает прочную связь тромбоцитов с субэндотелием сосудов; служит переносчиком одного их важнейших факторов свертывания – фактора VIII. Умеренное снижение уровня фактора фон Виллебранда или его высокомолекулярных олигомеров в плазме нарушает адгезию тромбоцитов и приводит к кровоточивости. Все типы болезни (кроме типа III) наследуются аутосомно-доминантно, причем все больные гетерозиготны.
Болезнь фон Виллебранда 2 типа (нормандская) далее делится на подтипы 2A, 2B, 2M и 2N. Мутантные белки при типах заболевания 2A, 2B и 2M имеют дефекты взаимодействия с тромбоцитами, тогда как мутантный белок при типе 2N имеет дефект связывания с F8. При 2 типе болезни фон Виллебранда уровень фактора близок к норме, но его активность снижена. При типе 2a существует дефицит высоко- и среднемолекулярных олигомеров фактора фон Виллебранда. Это связано с неспособностью секретировать высокомолекулярные олигомеры или с их разрушением в кровотоке. Мутации приводят к изменению небольшого участка молекулы в домене A-2. Уровни антигена фактора фон Виллебранда и связанного с ним фактора VIII обычно нормальны. При типе 2b уровень высокомолекулярных олигомеров тоже снижен, но обусловлено это избыточным связыванием фактора фон Виллебранда с тромбоцитами. Образующиеся агрегаты тромбоцитов быстро удаляются из кровотока, что приводит к легкой циклической тромбоцитопении. 3 тип болезни (самый редкий и тяжелый) наследуется аутосомно-рецессивно. Чаще всего оба родителя страдают легкой формой первого типа болезни. Больные с 3 типом заболевания могут быть как гомозиготами, так и смешанными гетерозиготами (то есть унаследовать от родителей разные дефекты). Характерны тяжелые кровотечения слизистых и отсутствие антигена или активности фактора фон Виллебранда; снижение активности фактора VIII иногда приводит к гемартрозам, подобно легким формам гемофилии. Описаны семьи с делециями больших участков гена фактора фон Виллебранда.
Дефицит прекалликреина (фактора Флетчера)
Заболевание вызвано мутациями в гене KLKB1, который кодирует калликреин плазмы крови (под воздействием калликреина плазмы на кининогены образуется брадикинин). Наследуется по аутосомно-рецессивному типу. Мутации в гене KLKB1 приводят к длительному активированному частичному тромбопластиновому времени, что свидетельствует о нарушении коагуляции. Болезнь может протекать бессимптомно или, в редких случаях, наблюдаются тромботические осложнения и повторяющиеся невынашивания беременности.
Дефицит протромбина (коагуляционного фактора II)
Заболевание наследуется аутосомно-рецессивно и встречается крайне редко. Вызывается мутациями в гене F2, которые приводят к двум основным типам заболевания: тип 1 известен как истинный дефицит протромбина или «гипопротромбинемия» и характеризуется пониженным содержанием протромбина в крови, составляющим 10% от нормы с сопутствующим снижением активности. У таких пациентов, начиная с момента рождения, наблюдаются тяжелые кровотечения, включая пуповинные кровотечения, гематомы, кровоподтеки, гематурию, кровотечения слизистых оболочек, гемартрозы, внутримозговое кровоизлияние, желудочно-кишечное кровотечение и меноррагию. Второй тип известен как «диспротромбинемия» и характеризуется синтезом дисфункционального протеина от нормального до пониженного. Симптомы кровотечений различны и зависят от количества оставшейся функциональной активности белка. У гетерозиготных носителей мутантных аллелей с уровнем протромбина в плазме 40–60% от нормы обычно отсутствует клиническая симптоматика, но у них могут возникать кровотечения при хирургическом вмешательстве и повышенная кровоточивость десен при чистке зубов, что подчеркивает важность диагностики носительства данных мутаций.
Rh-null синдром
Синдром вызван мутациями в гене RHAG, кодирующем связанный с резус-группой гликопротеин, который является аммонийтранспортирующим белком. Мутации в данном гене приводят к полному отсутствию Rh-антигенов, что выражается в анемии, снижении выживаемости эритроцитов, повышенной хрупкости эритроцитов, стоматоцитов и повышенному уровню фетального гемоглобина.
Врожденная амегакариоцитарная тромбоцитопения
Это редкий вид тромбоцитопении, связанный с поражением мегакариоцитопоэза, не поддающийся терапии иммуноглобулинами, стероидами и циклоспорином. Данное заболевание проявляется в младенчестве и вызывается мутациями в гене вируса миелопролиферативной лейкемии (MPL), которые ведут к возникновению двух типов заболевания. 1 тип вызывается нонсенс-мутациями и характеризуется полной потерей рецептора тромбопоэтина (полипептидного гормона, обеспечивающего созревание предшественников мегакариоцитов), что приводит к более тяжелым проявлениям – раннему началу тяжелой панцитопении, снижению активности костного мозга и очень низкому числу тромбоцитов. Второй тип вызывается нонсенс-мутациями, приводящими к снижению активности белка, и характеризуется более мягкими проявлениями с переходными увеличениями количества тромбоцитов до почти нормальных показателей в течение первого года жизни и недостаточностью костного мозга в 3-летнем возрасте и позже.
Наследственный сфероцитоз
Наследственный сфероцитоз объединяет группу заболеваний, которые характеризуются присутствием шаровидных эритроцитов (сфероцитов) в мазке крови и проявляются в виде анемии, желтухи, спленомегалии. На сегодняшний день описано пять типов наследственного сфероцитоза, которые вызваны мутациями в генах ANK1, SPTB, SPTBA, SLC4A1 и EPB42, кодирующими белки цитоскелета эритроцитов, преимущественно те, которые связывают цитоскелет с мембраной. В результате теряется часть мембраны эритроцита, уменьшается отношение площади поверхности к объему, и эритроцит превращается в микросфероцит. В большинстве случаев заболевание наследуется аутосомно-доминантно.
Хроническая лимфоцитарная лейкемия (ХЛЛ)
Заболевание характеризуется избыточной пролиферацией бластных форм лимфоидного ростка кроветворения, а клинически проявляется гиперплазией лимфатических узлов и селезенки. ХЛЛ редко встречается у азиатов, что свидетельствует о том, что в развитии болезни определенную роль играет генетический фактор. Наиболее распространенной формой ХЛЛ, составляющей почти три четверти всех случаев ХЛЛ, является В-клеточная лейкемия (В-лимфоцитарная лейкемия). Примерно в 1/3 случаев выявляются транслокации между одним из генов Т-клеточного рецептора и рядом онкогенов. Также, в 25% случаев обнаруживаются дефекты в регуляторной области гена TAL-I, участвующего в контроле гематопоэтического роста. Для зрелых В-клеточных лимфом характерна клональная перестройка генов иммуноглобулинов. При возникновении лимфомы на постфолликулярной стадии развития В-клеток также определяются мутации вариабельной области (гены V-области). В опухолях, развивающихся на стадии формирования центра размножения фолликулов, в В-клетках отмечаются продолжающиеся мутации генов V-области. Анализ генов иммуноглобулинов является надежным методом идентификации и дифференциации зрелых В-клеточных лимфом. Отсутствие соматических мутаций в вариабельном регионе генов тяжелой цепи иммуноглобулинов (IgVH) или наличие гена VH3.21 совпадает с менее благоприятным вариантом ХЛЛ. Экспрессия генов LAG3, LPL, ZAP-70 коррелирует с немутантным статусом IgVH и указывает на сокращение промежутка времени между сроком установления диагноза и началом специфической терапии. На предрасположенность к ХЛЛ указывают также дефекты в гене IRF4 – транскрипционном факторе, необходимом для развития клеток Т-хелперов (Th2), IL17-продуцирующих Th17 клеток и IL9-продуцирующих Th9 клеток. IRF4 ген априори является важным геном предрасположенности к ХЛЛ, поскольку является ключевым регулятором развития и пролиферации лимфоцитов. Кроме того, выявлено влияние полиморфизмов в генах серин/треонин протеинкиназы D2 (PKD2) и ядерного протеина SP140. Полиморфизмы гена TP53 также имеют неблагоприятный эффект. Определение статуса TP53 играет важную роль при прогнозировании чувствительности пациентов к некоторым лекарственным препаратам. Повышенная экспрессия в клетках пациентов с ХЛЛ антиапоптотического белка Mcl-1 семейства Bcl-2 ассоциируется с резистентностью к флударабину и хлорамбуцилу, а также со снижением выживаемости без прогрессии при терапии пентостатином, циклофосфамидом и ритуксимабом, что в совокупности указывает на возможную неэффективность стандартной химио- и химиоиммунотерапии.
Таким образом, возможность «обуздания» генетических секретов каждого из пациентов создает платформу для уникальной коррекции здоровья в противовес устранению отдельных симптомов заболевания. Четкая информация, полученная благодаря генетической диагностике, дает ключ к пониманию того, почему некоторые виды терапии оказываются неэффективными, а некоторые способы терапевтического воздействия являются наиболее подходящими.
Генетическое тестирование в любом возрасте, задолго до начала самого заболевания, позволяет не только идентифицировать уникальные особенности генотипа каждого человека, его генетическую индивидуальность, но и выявить предрасположенность к различным заболеваниям для своевременного проведения индивидуализированных профилактических мероприятий, направленных на предупреждение развития болезни. Именно генетические аспекты (изучение полиморфизма генов, ответственных за развитие и течение болезней крови) являются приоритетными в изучении гематологических заболеваний в настоящее время. Перспективными направлениями генетики в гематологии являются индивидуализированный подбор лекарственных препаратов на основании генетических маркеров, а также генно-инженерные методы лечения наследственной патологии системы крови.
Внедрение современных методов ДНК-тестирования в клиническую практику позволяет получить информацию о возможном риске развития у человека мультифакториальной патологии, а врач, принимая во внимание результаты молекулярно-генетического анализа, – разработать тактику упреждающей терапии предполагаемого заболевания, что позволит снизить заболеваемость, инвалидность и смертность населения. ДНК-тестирование с использованием современных молекулярно-генетических методов позволяет выявлять существующие пока только в геноме маркеры генетической предрасположенности к развитию гематологической патологии, сформировать группы «повышенного риска» по онкогематологическим заболеваниям с целью минимизации риска их развития.
Изучение генетической предрасположенности к гематологическим заболеваниям позволит лучше понять причины и механизмы развития патологического процесса, объяснить его разнообразие. Результаты ДНК-тестирования позволят разработать мероприятия по профилактике и своевременной диагностике заболеваний системы крови, будут способствовать разработке новых подходов к их лечению.
Детальніше
КАШЕЛЬ У ДЕТЕЙ
Кашель у детей – одна из наиболее частых причин обращения к врачу. Действительно, кашель является симптомом более 100 различных заболеваний. Но всегда ли кашель является проявлением патологического состояния? Что же такое кашель и нужно ли с ним бороться? Продолжая рубрику «Пограничных состояний», мы попытаемся дать дифференцированный подход к проблеме кашля с учетом возрастных особенностей детского организма. Надеемся, материалы этой рубрики будут вам полезны в повседневной врачебной деятельности
 Кашель как симптом – мультидисциплинарная проблема, поскольку с ним приходится сталкиваться в практической деятельности врачу-педиатру, семейному врачу, узкопрофильным специалистам: пульмонологам, фтизиатрам, отолагингологам, гастроэнтерологам, кардиологам.
Кашель как симптом – мультидисциплинарная проблема, поскольку с ним приходится сталкиваться в практической деятельности врачу-педиатру, семейному врачу, узкопрофильным специалистам: пульмонологам, фтизиатрам, отолагингологам, гастроэнтерологам, кардиологам.
Значение кашля для человека неоднозначно.
Кашель является:
а) важным защитно-приспособительным механизмом по очистке дыхательных путей от секрета и веществ, попавших в них извне;
б) фактором, способным удерживать пациента в сознании во время жизнеугрожающих аритмий и/или способствующим восстановлению нормального ритма сердечных сокращений;
в) фактором, способствующим распространению инфекции;
г) одним из наиболее частых симптомов, по поводу которых пациент обращается за медицинской помощью.
В силу того, что кашель является защитно-приспособительным рефлекторным актом, здоровые люди тоже могут и должны кашлять. Считается, что взрослый здоровый человек, в среднем, за сутки может в норме кашлянуть 11,5 раз. Как правило, люди, ведущие здоровый образ жизни, кашляют редко. Основным механизмом очистки дыхательных путей является мукоцилиарный транспорт, благодаря которому слизь и осевшие частички перемещаются в сторону глотки, откуда они откашливаются или проглатываются. Если кашель кратковременный и не повторяющийся ежедневно, то это может быть проявлением нормальной защитной реакции.
Нередко взрослые, а иногда и дети, могут использовать кашель как коммуникационный акт для привлечения внимания окружающих, для выражения отношения к услышанному и т. п. (покашливание, «тактичное покашливание», поперхнулся «от неожиданности», от возмущения).
Чаще кашель свидетельствует о наличии болезни. Поэтому врач должен уметь дифференцировать кашель как проявление нормальной реакции организма от симптома заболевания, а в этом случае уметь управлять кашлем, оптимизировать процесс удаления мокроты, используя рациональные методы терапии. В силу возрастных особенностей детского организма кашель у ребенка всегда должен вызывать настороженность врача.
Кроме того, если по той или иной причине кашель игнорируется или неправильно определена причина (этиология) кашля и, соответственно, назначается нерациональная и неэффективная терапия основного заболевания, то кашель становится далеко не безобидным симптомом и может приводить к различным осложнениям в результате резкого повышения внутригрудного давления (таблица 1). В числе наиболее тяжелых осложнений кашля можно назвать синкопальные состояния (беталепсия), аспирацию, спонтанный пневмоторакс, нарушения сердечного ритма, недержание мочи на фоне кашлевых приступов и др. Помимо этого, кашель нередко имеет нежелательные социальные последствия: вызывает стресс у самого больного, возникая в неподходящей ситуации, например, на концерте, уроке, экзамене, в транспорте и т. д., и негативные эмоции у окружающих, поскольку ассоциируется с такими опасными заболеваниями как туберкулез, что, в конечном итоге, приводит к социальной изоляции кашляющего человека. Вероятно, в связи с этим у людей с длительно существующим кашлем часто развивается депрессия. При оценке качества жизни таких пациентов взрослого возраста установлено, что влияние длительного кашля сравнимо с влиянием тяжелой хронической обструктивной болезни легких (ХОБЛ).
Мукоцилиарный транспорт является важнейшим механизмом, обеспечивающим санацию дыхательных путей, одним из основных механизмов системы местной защиты органов дыхания и обеспечивает необходимый потенциал барьерной, иммунной и очистительной функций респираторного тракта. Очищение дыхательных путей от чужеродных частиц и микроорганизмов происходит благодаря оседанию их на слизистых оболочках и последующему выведению вместе с трахеобронхиальной слизью.
Образование бронхиального секрета является одним из обязательных условий нормального функционирования бронхиального дерева. Бронхиальный секрет сложен по составу и является суммарным продуктом секреции слизистых и серозных клеток подслизистых желез, бокаловидных клеток, транссудации плазменных компонентов, метаболизма подвижных клеток и вегетирующих микроорганизмов, а также легочного сурфактанта. Обычно в бронхиальном секрете также обнаруживаются и клеточные элементы: альвеолярные макрофаги и лимфоциты. Трахеобронхиальная слизь в нормальных условиях обладает бактерицидным эффектом, т. к. содержит иммуноглобулины и неспецифические факторы защиты (лизоцим, трансферрин, опсонины и др.).
По физико-химической структуре бронхиальный секрет представляет собой многокомпонентный коллоидный раствор, состоящий из двух фаз: более жидкой (золь) и гелеобразной, нерастворимой. В растворимой фазе бактериального секрета содержатся электролиты, сывороточные компоненты, белки, биологически активные вещества, ферменты и их ингибиторы. Гель имеет фибриллярную структуру и образуется преимущественно за счет местно синтезированных макромолекулярных гликопротеиновых комплексов муцинов, сцепленных дисульфидными мостиками. Золь покрывает апикальные поверхности мукоцилиарных клеток. Именно в этом слое реснички мерцательного эпителия совершают свои колебательные движения и передают свою кинетическую энергию наружному слою – гелю.
Скорость мукоцилиарного транспорта у здорового человека колеблется от 4 до 20 мм в минуту, в норме за сутки транспортируется от 10 до 100 мл бронхиального секрета. Реснитчатый эпителий выводит частицы, осевшие в воздухоносных путях, в течение нескольких часов, в то время как частицы, достигшие альвеол, удаляются через несколько дней или месяцев. В последнем случае имеет значение их фагоцитоз альвеолярными макрофагами. Очищающая функция эпителия может быть усилена кашлевыми толчками, удаляющими избыток слизи под давлением до 300 мм рт. ст. и при скорости потока воздуха 5–6 л/с. Слизь, выброшенную кашлевыми толчками, маленькие дети обычно проглатывают.
Что же представляет собой кашель?
Кашель представляет собой произвольный или непроизвольный (рефлекторный) толчкообразный форсированный звучный выдох. Афферентная часть рефлекторной дуги кашлевого рефлекса образована волокнами тройничного, языкоглоточного, верхнего гортанного и блуждающего нервов.
Кашлевой рефлекс обычно инициируется стимуляцией чувствительных нервных окончаний ротовой полости, придаточных пазух носа, гортани, голосовых связок, глотки, наружного слухового прохода, евстахиевой трубы, трахеи и ее бифуркации, мест деления бронхов (бронхиальные шпоры), плевры, перикарда, диафрагмы, дистальной части пищевода и желудка.
Кашлевые рецепторы, локализующиеся в дыхательных путях, представлены двумя типами нервных окончаний:
- быстро адаптирующиеся или ирритантные рецепторы, реагирующие на механические, термические и химические раздражители и расположенные в проксимальных отделах респираторного тракта;
- С-волокна или С-рецепторы, стимулируемые различными провоспалительными медиаторами (простагландины, брадикинины, субстанция Р и др.) и локализующиеся более дистально.
При этом наиболее чувствительными рефлексогенными зонами в дыхательных путях являются:
- гортанная поверхность надгортанника;
- передняя межчерпаловидная поверхность гортани;
- область голосовых связок и подсвязочного пространства;
- бифуркация трахеи и места ответвления долевых бронхов.
В то же время, по направлению к дистальным отделам бронхиального дерева плотность кашлевых рецепторов уменьшается; одновременно с этим они становятся более чувствительными к раздражающим воздействиям, вызывающим кашель.
Возникающий при раздражении рефлексогенных зон импульс передается через афферентные волокна в кашлевой центр, расположенный в продолговатом мозге. Рефлекторная дуга замыкается эфферентными волокнами возвратного гортанного, диафрагмального и спинномозговых нервов, идущих к мышцам-эффекторам – грудной клетки, диафрагмы и брюшного пресса.
Кашель начинается с глубокого вдоха, после которого закрывается голосовая щель, и сокращаются дыхательные мышцы. За счет синхронного напряжения дыхательной и вспомогательной мускулатуры при закрытой голосовой щели нарастает внутригрудное давление, сужаются трахея и бронхи. При открытии голосовой щели резкий перепад давления создает в суженных дыхательных путях стремительный поток воздуха (форсированный толчкообразный выдох), увлекающий за собой слизь и инородные частицы. Содержимое из легких при кашле не поступает через нос, так как во время кашля носовую полость закрывает мягкое небо.
Если перистальтические движения мелких бронхов и деятельность реснитчатого эпителия крупных бронхов и трахеи не обеспечивают необходимый дренаж, развивается кашель. Следовательно, кашель – это второй защитный механизм, направленный на восстановление проходимости дыхательных путей.
Однако кашель выполняет защитную функцию только при определенных реологических свойствах мокроты и отсутствии препятствия для ее эвакуации.
Говоря о проблеме кашля в педиатрической практике, нельзя не учитывать анатомо-физиологические особенности дыхательного тракта у детей, которые накладывают отпечатки на формирование клинических проявлений и течение заболевания в целом, в том числе влияют на характер кашля:
- преобладание более плотного гелевого слоя бронхиального секрета над золевым, его повышенная вязкость вследствие высокого содержание сиаловой кислоты;
- относительно большее, чем у взрослых, количество бокаловидных клеток на единицу площади слизистой оболочки и ее обильная васкуляризация;
- трахея и бронхи имеют относительно и абсолютно узкий просвет, движение в бронхиолах более медленное, чем в бронхах, что способствует застою слизи и обусловливает частые закупорки слизистыми пробками, частые бронхиолиты у детей грудного возраста;
- вследствие слабого развития дыхательной мускулатуры, мышечных и эластичных волокон бронхиального каркаса отмечаются слабость и неэффективность кашлевых толчков у детей младшего возраста.
В совокупности эти особенности обусловливают склонность к гиперкринии, отеку и сужению просвета дыхательных путей при воспалении. Чем младше ребенок, тем менее эффективный у него кашель и тем труднее ему откашлять мокроту, что предрасполагает к присоединению вторичной микрофлоры и развитию осложнений.
Диагностика кашля
В определении причин кашля важную роль играет профессионально собранный анамнез. В первую очередь необходимо выяснить следующее:
- Как давно появился кашель?
- Частота, тембр (высота) и интенсивность кашля. Меняются ли эти характеристики кашля на протяжении суток (исчезает во сне или когда ребенок успокаивается)?
- Чем он был инициирован: приемом пищи, во время игры ребенка мелкими предметами (аспирация)?
- Нет ли лихорадки? Имеются ли выделения из носа, частые покашливания (ринит, синусит), изжога или отрыжка (гастроэзофагеальный рефлюкс)?
- Предшествовала ли кашлю респираторная инфекция?
- Характерны ли сезонные обострения? Бывают ли приступы удушья или свистящего дыхания? Отмечаются ли аналогичные симптомы у родственников (бронхиальная астма)?
- Отходит ли во время кашля мокрота? Если да, то в каком количестве и какого цвета?
- Сопровождается ли кашель одышкой, периферическими отеками, цианозом?
- Нет ли других заболеваний или факторов риска (курение, неблагоприятные факторы окружающей среды)?
- Принимает ли больной препараты, прием которых может сопровождаться кашлем (ингибиторы АПФ)?
Классификация кашля
- По продолжительности (острый – до 3 недель, подострый – от 3 до 8 недель, хронический – более 8 недель);
- По характеру (продуктивный, непродуктивный – влажный, сухой);
- По времени появления кашля (утренний, вечерний, ночной);
- По ритму (отдельный кашлевой толчок, ряд следующих друг за другом кашлевых толчков, приступообразный кашель);
- По тембру (поверхностный, лающий, битональный, сиплый, беззвучный).
Наиболее частыми причинами острого и подострого кашля у детей являются: острая респираторная вирусная инфекция, вызванная респираторно-синцитиальным вирусом, вирусами гриппа, парагриппа, аденовирусом, респираторным корона-вирусом, метапневмавирусом, бокавирусом, риновирусом; острый и рецидивирующий бронхит; коклюш; пневмония, плеврит; заболевания верхних дыхательных путей (постназальный затек при ринитах/риносинуситах, ринофарингитах, аденоидитах, ларингиты/лагинготрахеиты); аспирация инородного тела; различные раздражающие вещества (пыль, дым, газы). Острый кашель может быть симптомом сердечной недостаточности, тромбоэмболии ветвей легочной артерии (встречаясь едва ли не в половине случаев, у отдельных больных он может оказаться единственным респираторным симптомом).
В некоторых случаях у ранее здоровых лиц непродуктивный кашель после разрешения инфекции верхних дыхательных путей (особенно РС-инфекции, гриппа, аденовируса) может сохраняться значительный промежуток времени, так называемый постинфекционный кашель. После перенесенного острого бронхита кашель также может сохраняться в течение нескольких недель вследствие повышенной чувствительности бронхов. Нераспознанный коклюш может быть причиной длительного кашля, характеризующегося выраженностью и мучительным характером.
В рекомендациях Американской коллегии специалистов по заболеваниям органов грудной клетки (American College of Chest Physicians – ACCP), кашель после перенесенной острой инфекции респираторного тракта считается постинфекционным при его продолжительности 3–8 недель. Если же кашель длится >8 нед, необходимо рассматривать другой диагноз.
По данным зарубежной литературы хронический кашель как единственная жалоба отмечается в 10–38% случаев всех обращений взрослых пациентов к специалистам. Причинами хронического кашля у детей могут быть бронхиальная астма, туберкулез, муковисцидоз, интерстициальные заболевания легких, пороки развития бронхов и легких, опухоли, а также пороки сердца, перикардит, сердечная недостаточность, гастроэзофагеальная болезнь, психогенный и рефлекторный кашель. Эпидемиологическая ситуация, сложившаяся в Украине в последние годы, требует от врачей настороженности в отношении туберкулеза и оппортунистических инфекций дыхательных путей при ВИЧ/СПИДе. У пациентов с хроническим кашлем наблюдается повреждение эпителия дыхательных путей наряду с воспалительной инфильтрацией. Обнажение окончаний чувствительных нервов при повреждении эпителия может также способствовать усилению кашлевого рефлекса при воздействии экзогенных и эндогенных факторов. При хронических болезнях органов дыхания длительный, постоянный кашель может усиливаться или ослабевать в отдельные периоды времени, но принципиально важно, что ребенок практически постоянно кашляет.
Существенным усугубляющим фактором является воздействие поллютантов или ирритантов. У взрослых и детей школьного возраста хронический ночной кашель связан с уровнем загрязнения воздуха.
Проживание вблизи трасс с интенсивным движением может ассоциироваться с симптомами бронхиальной астмы и хроническим кашлем. У £10% детей дошкольного и раннего школьного возраста отмечается стойкий хронический кашель без одышки, не связанный с простудой. Он может быть ассоциирован с такими факторами окружающей среды как влажность в доме и загрязнение воздуха, что, в свою очередь, тесно связано с социально-экономическим статусом семьи.
Одной из распространенных причин упорного кашля является курение, при этом прекращение курения ведет к кратковременному повышению чувствительности кашлевого рефлекса. Доказано, что курение родителей ассоциировано с повышением частоты случаев хронического кашля у детей: данная проблема отмечается у 50% детей в возрасте до 11 лет в случае, если курят оба родителя.
Продуктивность кашля
По характеру отделения секрета кашель может быть продуктивный (влажный) или непродуктивный (сухой). Наиболее мучительным бывает сухой (непродуктивный) кашель. Такой кашель может сохраняться несколько дней, но в некоторых случаях продолжается и в течение более длительного отрезка времени.
Непродуктивный кашель развивается при химическом, механическом или термическом раздражении дыхательных путей (дым, пыль), атрофических процессах, часто полностью прекращается после устранения раздражителя. В качестве механических причин могут выступать инородные тела; давление на воздухоносные пути увеличенных медиастинальных лимфатических узлов, опухолей, аневризматически расширенной аорты или дупликатуры дуги аорты; а также подтягивание легочной паренхимы при фиброзирующих процессах (ателектазы, фиброз). Сухой, часто приступообразный, с металлическим оттенком, иногда «ревущий» (honking), кашель может развиваться при патологическом раздражении рецепторов кашлевого центра продолговатого мозга (кашель центрального генеза – опухоли, эпилепсия и др. органические поражения ЦНС). Сухой кашель у ребенка младшего возраста с длительным (месяцами) изменением голоса (осиплость) может указывать на папилломатоз гортани.
Сухой непродуктивный кашель отмечается также при инфекционных и аллергических заболеваниях верхних дыхательных путей, бронхиальной астме, интерстициальных заболеваниях легких (фиброзирующие альвеолиты, пневмокониозы, саркоидоз). Непродуктивный кашель отмечается при кори, коклюше, паракоклюше, респираторном хламидиозе и микоплазмозе.
Сухой непродуктивный кашель может быть проявлением гастроэзофагеальной рефлюксной болезни или миграции круглых гельминтов. Причиной хронического непродуктивного кашля могут быть и врожденные пороки сердца вследствие компрессии бронхов повышенным давлением в легочной артерии, расширением левого предсердия или сужением периферических дыхательных путей из-за легочного отека. К ним относятся открытый артериальный проток, стеноз легочной артерии, тетрада Фалло.
Продуктивность или непродуктивность кашля может зависеть от многих причин. Это фазность течения заболевания (при пневмонии кашель в течение некоторого времени остается сухим, при хроническом бронхите, бронхиальной астме (БА) нарастание обструкции может сопровождаться прекращением экспекторации мокроты), локализация процесса (например, разный по продуктивности кашель в зависимости от локализации туберкулезного процесса в легких – в паренхиме легкого, бронхах, плевре).
Анализируя характер продуктивного кашля и особенности мокроты, следует обращать внимание на:
- кашель с отделением светлой мокроты (желтого/зеленого цвета в периоды обострения) – при хроническом бронхите, муковисцидозе;
- гнойную мокроту, расслаивающуюся при стоянии – нагноившиеся бронхоэктазы, абсцесс легкого;
- гнойную зловонную мокроту – при гангрене легкого;
- кровянистую мокроту – при инфаркте легкого, туберкулезе легких, бронхогенной карциноме, бронхоэктазии, застойных явлениях в малом круге кровообращения;
- розовую пенистую мокроту – при гемодинамическом отеке легких.
В силу анатомо-физиологических особенностей у грудных детей после перенесенного острого или обструктивного бронхита очень часто отмечается сохранение гиперсекреции слизи при повышении кашлевого порога, что обуславливает редкий влажный кашель в течение длительного времени (4 недели и более). Отличительная особенность такого кашля – наличие «хрипотцы» – клокочущих звуков в грудной клетке, слышимых на расстоянии, которые исчезают после кашля и возникают вновь по мере накопления мокроты. Мокрота из трахеи и гортани у грудных детей эвакуируется более редкими кашлевыми толчками, когда просвет бронхов будет почти полностью перекрыт. У таких детей кашель при давлении на трахею (или шпателем на корень языка) вызывается с трудом. Кашель, связанный с гиперсекрецией, постепенно стихает – как по частоте, так и по интенсивности.
Может иметь место и ложная продуктивность – например, при постназальном затеке у больных с ринитами и/или синуситами, аденоидитами откашливается назальная слизь. Когда речь идет о продуктивном кашле (с отхождением различной мокроты: слизистой, слизисто-гнойной, гнойной), следует иметь в виду, что в части случаев он может оказаться несостоятельным (неэффективным), т. е. не выполняющим в достаточной мере дренажную функцию по следующим причинам:
- Недостаточно выраженный кашлевой рефлекс: недостаточная моторика бронхов и недостаточная «подача» мокроты в зону кашлевого рефлекса; снижение возбудимости кашлевого центра, что зависит от возраста (грудные дети, старики) или может быть связано с интоксикацией (гиперкапния, токсическое угнетение центральной нервной системы при инфекциях), а также наблюдаться во время наркоза, глубокого сна; снижение чувствительности рецепторов в бронхах – местная анестезия (например, после ингаляций парами ментола); дегенерация нервных окончаний в результате хронического воспаления.
- Слишком большая вязкость мокроты.
- Малая мощность воздушной струи во время кашлевого толчка, обусловленная ригидностью грудной клетки, малой податливостью легких или, чаще всего, нарушениями бронхиальной проходимости.
- Недостаточно глубокое дыхание (продвижение секрета в бронхах пропорционально глубине дыхания).
Кроме того, в определенных клинических ситуациях (при переломе ребер, других травматических повреждениях грудной клетки, после перенесенных хирургических вмешательств на органах грудной клетки и брюшной полости и др.) больные произвольно «противятся» кашлю.
Продолжение в следующем номере.
Детальніше
АУТОВОСПАЛИТЕЛЬНЫЕ СИНДРОМЫ КАК ПРИЧИНА РЕЦИДИВИРУЮЩЕЙ ЛИХОРАДКИ У ДЕТЕЙ
Дифференциальная диагностика при лихорадке неясного генеза всегда представляет значительные трудности для клинициста. Как известно, причинами такой лихорадки могут быть инфекционные, онкологические, аутоиммунные заболевания. Кроме того, в качестве возможных причин длительной лихорадки, особенно при рецидивирующем ее характере, рассматриваются так называемые периодические синдромы
Впервые термин «периодическая болезнь» появился в 1948 году для обозначения пациентов с повторными эпизодами лихорадки, появляющимися в детском возрасте и сохраняющимися на протяжении многих лет или десятилетий. Позднее стало понятно, что «периодическая болезнь» – это неоднородная группа различных по своей природе заболеваний, общей особенностью которых является наличие приступов лихорадки, сопровождающихся другими признаками острого воспаления, наследственный характер, начало в детском возрасте. В 80-е годы ХХ столетия благодаря успехам генетики и молекулярной биологии список наследственных синдромов с периодической лихорадкой расширился до четырех заболеваний, а затем до восьми.
Изучение природы периодических синдромов лихорадки показало, что в основе их лежит гиперактивация неспецифического (врожденного) иммунитета, приводящая к развитию спонтанного системного воспаления, с лихорадкой и вовлечением многих органов, но при этом отсутствуют аутоантитела или какие-либо другие признаки аутоиммунитета. На основании этого наблюдения был предложен термин «аутовоспалительные заболевания/синдромы», и со временем учение об «аутовоспалительных синдромах» оформилось в отдельный раздел ревматологии.
В настоящее время аутовоспалительные синдромы (АВС) являются предметом активного изучения во всем мире. Если изначально в эту группу были включены лишь заболевания с генетическими дефектами, обусловливающими дисрегуляцию врожденного иммунитета, то сейчас список АВС дополнен идиопатическими, а также полигенными заболеваниями, имеющими черты аутовоспаления (табл. 1). Единой классификации АВС нет, список этих заболеваний постоянно пополняется, уточняется и расширяется. В настоящее время он насчитывает более 25 нозологических форм.
Несмотря на то, что АВС являются достаточно редкими заболеваниями, практически все они начинаются в детском возрасте, поэтому любой педиатр имеет шанс столкнуться с ними. Характерные черты АВС – периодическая лихорадка, сочетающаяся с другими признаками воспаления (кожные проявления, мышечно-суставной синдром, серозиты, повышение уровней острофазовых показателей) при отсутствии инфекции и признаков аутоиммунитета (аутоантител, активации аутоспецифических клеток). При некоторых АВС развивается амилоидоз с последующей хронической почечной недостаточностью. В настоящее время благодаря появлению генно-инженерных биологических препаратов наметился существенный прогресс в лечении АВС, поэтому своевременная их диагностика очень важна. В данной статье мы рассмотрим наиболее часто встречающиеся АВС.

Семейная средиземноморская лихорадка (Familial Mediterranean Fever – FMF)
Это наследственное заболевание, встречающееся среди лиц, принадлежащих к средиземноморским этническим группам (евреи-сефарды, арабы, турки, армяне, итальянцы), характеризующееся короткими приступами лихорадки, вызванными индуцированным нейтрофилами воспалением серозных оболочек с постепенным развитием амилоидоза почек.
Генетическая основа заболевания – мутации в гене MEFV, локализованном в коротком плече 16 хромосомы и кодирующем синтез регуляторного белка пирина (или маренострина). Пирин является противовоспалительным агентом, экспрессирующимся, главным образом, в нейтрофилах и цитокин-активированных моноцитах. Он связывает белок ASC, что, в свою очередь, предотвращает активацию каспазы-1. Каспаза-1 повышает синтез ИЛ-1, активирует нуклеарный фактор каппа В и ИЛ-4-индуцированный апоптоз клеток в ответ на воздействие ЛПС.
Таким образом, у пациентов с FMF имеется аномально повышенная чувствительность к бактериальному эндотоксину, когда транзиторная бактериемия (в норме – абсолютно безвредное состояние) приводит к значительному повышению синтеза ИЛ-1a с развитием системного воспалительного ответа. Другими индукторами ИЛ-1a при FMF могут быть чрезмерные физические нагрузки, эмоциональное напряжение, изменение гормонального фона (у женщин). Под действием провоспалительных цитокинов в печени повышается синтез белков острой фазы воспаления, в т. ч. сывороточного амилоида А, продукты деградации которого откладываются во внутренних органах. Развитие амилоидоза почек – наиболее серьезное осложнение FMF, определяющее прогноз заболевания.
Большинство случаев FMF обусловлено 5 мутациями гена MEFV (M694V, V726A, M680I, M694I, E148Q). Тип наследования – аутосомно-рецессивный, при некоторых мутациях – со сниженной пенетрантностью.
FMF – наиболее часто встречающийся наследственный АВС. Практически каждый четвертый представитель средиземноморских этнических групп является носителем одной из мутаций гена MEFV.
Первые признаки заболевания чаще всего появляются в детском возрасте: у 90% пациентов – до 20 лет жизни, у 60% – до 10-летнего возраста. Чем раньше манифестирует заболевание, тем тяжелее оно протекает. Соотношение лиц мужского и женского пола при FMF составляет 1,5-2:1, что может быть связано со сниженной пенетрантностью мутантных генов у женщин. Кроме того, на течение болезни у женщин влияют половые гормоны – нередко приступы болезни возникают во время менструаций, отсутствуют во время беременности и возобновляются после родов. Риск развития амилоидоза почек выше у мужчин.
Клинически семейная средиземноморская лихорадка проявляется повторными приступами острой боли и высокой лихорадки, которые длятся от 12 часов до 4 дней. Боль обычно имеет одну-две из следующих локализаций: живот, грудь, суставы, мышцы, мошонка, кожа.
Наиболее часто (82–98% случаев) отмечается абдоминальная боль по типу «острого живота». Присутствует напряжение мышц передней брюшной стенки, положительные симптомы раздражения брюшины, ослабление перистальтики. У одной трети пациентов отмечают увеличение селезенки. У некоторых пациентов боль в животе может иметь место и в межприступном периоде, что связано с формированием спаек брюшины вследствие рецидивирующего воспаления. Сообщалось о высокой частоте мутаций MEFV среди арабских и еврейских детей с функциональной абдоминальной болью, которая не соответствовала классическим проявлениям FMF.
У 70% пациентов во время приступов лихорадка сочетается с поражением суставов, обычно в виде кратковременного недеструктивного острого моноартрита. Чаще всего вовлекаются крупные суставы нижних конечностей. Приблизительно у 1% пациентов артрит – единственное проявление FMF.
Миалгии – нередкий симптом при FMF. Синдром фебрильной миалгии – тяжелый, болезненный, дезадаптирующий приступ, длящийся несколько недель и купирующийся только глюкокортикоидами.
У 40% пациентов в период приступа отмечаются симптомы плеврита: односторонняя боль в груди, усиливающаяся при дыхании, кашель, одышка, поверхностное частое дыхание. У гомозигот по мутации M694V плевриты встречаются значительно чаще, чем у гомозигот по мутации V726A или любой другой комбинации мутаций.
Наиболее типичное поражение кожи при FMF – эритема на нижних конечностях, напоминающая рожистое воспаление. Отечные гиперемированные болезненные участки кожи 10–15 см в диаметре обычно локализуются ниже колена на передней или задней поверхности ноги, с одной стороны или симметрично. Кроме того, васкулит является важным, но все еще не широко признанным признаком FMF, который может предшествовать классическим проявлениям болезни. Известно, что геморрагический васкулит Шенляйн–Геноха и узелковый периартериит у пациентов с FMF обнаруживают чаще, чем в общей популяции.
Воспаление оболочек яичка с клиникой «острой мошонки» отмечается примерно у 5% пациентов. Поражение, как правило, одностороннее, характеризуется болезненностью, отечностью и гиперемией мошонки на пораженной стороне, проходит самостоятельно по окончании приступа.
Другие, редкие, но хорошо описанные проявления FMF – перикардит, менингит, головная боль во время приступов и бесплодие у женщин (из-за спаечного процесса в брюшной полости).
В межприступном периоде пациенты здоровы.
Основное осложнение FMF – амилоидоз почек. При отсутствии ранней и непрерывной терапии колхицином, амилоидоз почек может развиться уже в течение нескольких лет с момента появления первых симптомов заболевания с формированием нефротического синдрома и хронической почечной недостаточности. При FMF имеет место АА-амилоидоз (вторичный), когда во внутренних органах откладывается амилоид А, являющийся продуктом распада сывороточного белка SAA. Это белок острой фазы воспаления, который вырабатывается в печени под действием ИЛ-1. У евреев-сефардов с FMF амилоидоз почек развивается более чем в 90% случаев. Пациенты других этнических групп менее подвержены этому осложнению. Развитие амилоидоза почек главным образом характерно для гомозигот по мутации M694V, однако есть данные, что он может встречаться и при сочетании этой мутации с другими генотипами, при которых отмечается не такое тяжелое течение заболевания. Более того, амилоидоз почек можно обнаружить у пациентов с бессимптомным течением FMF, т. е. при отсутствии приступов серозитов.
Во время приступов лихорадки в крови пациентов отмечается нейтрофильный лейкоцитоз со сдвигом формулы влево, повышение СОЭ и других острофазовых показателей (уровня С-реактивного белка, SAA-протеина, фибриногена, гаптоглобина, С3- и С4-фракций комплемента). Возможно развитие анемии хронического воспаления. В общем анализе мочи во время лихорадочных эпизодов может отмечаться транзиторная гематурия и небольшая протеинурия. Если же уровень экскреции белка превышает 0,5 г/сут., то это свидетельствует в пользу амилоидоза почек.
До 1997 года диагноз семейной средиземноморской лихорадки был сугубо клиническим. С открытием гена FMF стало возможным подтверждение диагноза молекулярно-генетическими методами.
Дифференциальная диагностика при семейной средиземноморской лихорадке проводится с острым аппендицитом, острыми процессами в малом тазу, панкреатитом, порфирией, наследственным ангионевротическим отеком, геморрагическим васкулитом, узелковым периартериитом, пневмонией, плевритом, септическим артритом, ЮРА с системным началом, инфекционным перикардитом, амилоидозом другого происхождения, другими наследственными синдромами периодической лихорадки и аутовоспалительными синдромами, циклической нейтропенией, синдромом «острой мошонки» (перекрут яичка, эпидидимит, орхит).
Диагностические критерии семейной средиземноморской лихорадки (Livneh A. et al., 1997)
Большие критерии:
- рецидивирующие эпизоды лихорадки с перитонитом, синовиитом или плевритом;
- AA-амилоидоз в отсутствие других предрасполагающих к нему заболеваний;
- позитивный ответ на постоянную терапию колхицином.
Малые критерии:
- рецидивирующие эпизоды лихорадки;
- рожеподобная эритема;
- FMF у родственников первой степени родства.
2 больших критерия или 1 большой и 2 малых – диагноз убедительный;
1 большой критерий и 1 малый критерий – диагноз вероятный.
Основной метод лечения FMF – постоянный прием колхицина. Приблизительно у 65% пациентов на фоне приема колхицина симптомы заболевания полностью проходят, у 30% наблюдается существенное улучшение и у около 5% больных FMF лечение колхицином неэффективно. Поскольку терапия колхицином позволяет не только устранить приступы лихорадки и серозитов, но и предотвратить развитие почечного амилоидоза, пожизненный прием препарата рекомендуется всем больным FMF, независимо от тяжести течения, включая пациентов, не отвечающих на лечение. Начальная доза препарата — 1 мг/сут. (для детей младше 5 лет — 0,5–0,6 мг/сут.). Если терапевтический эффект не достигнут, доза колхицина может быть увеличена. Максимальная доза — 3 мг/сут.
В терапии начавшегося приступа колхицин неэффективен. С целью купирования симптомов лихорадочных атак назначают НПВС. При синдроме фебрильной миалгии эффективны только глюкокортикоиды.
Периодический синдром, ассоциированный с рецептором ФНО (Tumor necrosis factor (TNF) receptor-associated periodic syndrome – TRAPS) или семейная ирландская лихорадка
Это аутосомно-доминантно наследуемое заболевание, характеризующееся приступами лихорадки, абдоминальной боли, интенсивной миалгии и болезненной эритемы на туловище или конечностях, обычно длящимися дольше 1 недели. TRAPS-синдром впервые был описан в 1982 г. в большой ирландско-шотландской семье.
Заболевание вызвано мутациями в гене TNFRSF1A, кодирующем рецептор ФНО. Под воздействием ФНОa внеклеточно расположенная часть TNFRSF1A подвергается расщеплению с последующим слущиванием с мембраны клетки. В результате увеличивается количество растворимых рецепторов ФНО, которые связывают данный цитокин во внеклеточной среде, предотвращая тем самым связывание ФНО рецепторами, расположенными на мембранах клеток, а значит и последующие провоспалительные эффекты.
При большинстве мутаций TNFRSF1A уменьшается количество растворимых рецепторов ФНО, что способствует индукции и поддержанию воспалительного ответа. Дефект слущивания рецепторов ФНО лишь частично объясняет патогенез TRAPS-синдрома, так как при некоторых мутациях данный процесс не нарушен. Учитывая, что применение при данном заболевании этанерцепта – антагониста ФНО, сопровождается быстрым наступлением стойкой ремиссии, предполагается, что и сам ФНО играет определенную роль в развитии этого синдрома.
Большинство пациентов с TRAPS-синдромом имеет североевропейское происхождение. Несмотря на то, что первоначально заболевание было описано у ирландцев и шотландцев, мутации TNFRSF1A были найдены и у пациентов других этнических групп, включая французов, бельгийцев, голландцев, афроамериканцев, арабов, евреев и многих других. Соотношение мужчин и женщин среди больных TRAPS-синдромом составляет 3:2. Возраст начала заболевания может быть самым различным – от 2 недель до 53 лет (в среднем – 3 года). Возраст появления первых симптомов может значительно варьировать даже внутри одной семьи.
Частота и длительность воспалительных атак при TRAPS-синдроме могут быть различными. В среднем, они происходят каждые 6 недель и длятся дольше 1 недели. При этом у многих пациентов болевой синдром может сохраняться и между лихорадочными приступами.
Асептическое воспаление серозных оболочек брюшной и грудной полостей, сопровождающееся болью в животе и груди, в период лихорадочного приступа встречается у 90% и 60% пациентов соответственно.
Как правило, отмечаются артралгии крупных суставов, но артриты возникают редко.
Также характерным признаком атак при TRAPS-синдроме является односторонний или двусторонний конъюнктивит, сопровождающийся сильной болью, а также периорбитальный отек.
Миалгии – патогномоничный симптом для TRAPS-синдрома. Обычно приступы начинаются именно с мышечных болей, при этом в течение приступа миалгии мигрируют в дистальном направлении.
Приблизительно у 84% пациентов отмечается болезненная, мигрирующая эритема, обычно локализующаяся над областями миалгий и сохраняющаяся в течение 4–21 дня.
У пациентов мужского пола в период атак может отмечаться боль в области мошонки.
Диагностические критерии TRAPS-синдрома (Hull K. M. et al., 2002):
- Наличие в течение не менее чем 6 месяцев повторных приступов лихорадки, сочетающейся с:
- болями в животе;
- мигрирующей миалгией;
- мигрирующей эритемой;
- конъюнктивитом, периорбитальным отеком;
- болью в груди;
- артралгиями или артритом.
- Длительность эпизодов более 5 дней.
- Симптомы уменьшаются при лечении глюкокортикоидами, но не колхицином.
- Аналогичные симптомы у членов семьи.
- Любая этническая принадлежность.
Триггерами воспалительных атак при TRAPS-синдроме могут быть физическое или эмоциональное напряжение, физическая травма.
Как и при других АВС, в период приступов повышаются уровни острофазовых показателей (СОЭ, СРБ, SAA, фибриноген, гаптоглобин, ферритин). СОЭ может оставаться повышенной и в межприступном периоде. В общем анализе крови можно выявить анемию хронического заболевания, лейкоцитоз, тромбоцитоз. Кроме того, при TRAPS-синдроме может быть повышен уровень иммуноглобулина D (IgD), но не выше 100 ед./мл. Типичным является снижение уровня растворимого рецептора ФНО (TNFRSF1A) в сыворотке крови, как в период приступа, так и между атаками. Нормальный уровень растворимого TNFRSF1A не исключает диагноз. Окончательно диагноз подтверждается генетическими исследованиями, обнаруживающими мутации в гене TNFRSF1A.
Основным осложнением TRAPS-синдрома, определяющим его прогноз и продолжительность жизни пациентов, является АА-амилоидоз.
Тяжесть течения заболевания, так же, как и риск развития амилоидоза, определяется характером мутаций. Всего на сегодняшний день описано 58 мутаций в гене TNFRSF1A. Наиболее неблагоприятными являются миссенс-мутации, приводящие к замене цистеина во внеклеточной части рецептора. При мутациях, затрагивающих нецистеиновые остатки, снижается пенетрантность клинического фенотипа (82% против 93% при заменах цистеина), а риск амилоидоза уменьшается до 2% (против 24% при цистеиновых мутациях).
Дифференциальный диагноз включает другие аутовоспалительные синдромы с периодической лихорадкой, особенно FMF. Нередко также приходится проводить дифференциальную диагностику с острым перитонитом. Значительное число пациентов с TRAPS-синдромом подвергается диагностической лапаротомии и аппендэктомии.
Лечение лихорадочных приступов при TRAPS-синдроме включает применение НПВС, которые способствуют нормализации температуры тела, но неэффективны в отношении скелетно-мышечных и абдоминальных симптомов. Глюкокортикоиды уменьшают выраженность симптомов заболевания у большинства пациентов. Ни НПВС, ни глюкокортикоиды не влияют на частоту приступов.
Получены предварительные результаты эффективности этанерцепта в лечении и профилактике лихорадочных атак при TRAPS-синдроме. Этанерцепт – димерный рекомбинантный белок, состоящий из внеклеточной части рецептора ФНОa типа 2, связанного с Fc-фрагментом IgG1. Этот белок связывает ФНО во внеклеточном пространстве, уменьшая тем самым его биологические эффекты. В исследованиях было показано, что подкожное введение этанерцепта в стандартных дозах два раза в неделю уменьшает частоту, длительность и тяжесть приступов. Кроме того, этанерцепт препятствует развитию амилоидоза у пациентов с TRAPS-синдромом. С этой целью препарат, по всей видимости, нужно принимать пожизненно. Хотя полученные предварительные результаты весьма обнадеживают, требуется проведение долгосрочных двойных слепых исследований для того, чтобы точнее определить роль терапии этанерцептом в клиническом менеджменте TRAPS-синдрома и оценить ее эффект в отношении АА-амилоидоза.
ГиперIgD-синдром
Гипер IgD-синдром (Hyper IgD Syndrome – HIDS), известный также как дефицит мевалонаткиназы (mevalonate kinase deficiency – MKD) – аутосомно-рецессивное заболевание, характеризующееся повторными приступами лихорадки с ознобами и недомоганием, сопровождающимися повышением сывороточного уровня IgD. Заболевание впервые было описано в 1984 г. у 6 пациентов – голландцев по происхождению.
Гипер IgD-синдром связан с мутациями в гене мевалонаткиназы, которые приводят к снижению ферментативной активности. Мавалонаткиназа обнаруживается, главным образом, в пероксисомах и катализирует ранние этапы мевалонового пути синтеза холестерина и других стеролов (витамина Д, желчных кислот). Другими конечными продуктами являются изопреноиды. Механизм развития воспалительных атак при дефиците мевалонаткиназы до конца неясен. Известно лишь, что некоторые изопреноиды связаны с апоптозом, который играет важную роль в подавлении воспалительного ответа. Было показано, что циркулирующие лимфоциты у пациентов с HIDS отличаются сниженным апоптозом.
Большинство случаев HIDS описано у западных европейцев – французов и голландцев. Недавно было выяснено, что частота мутаций мевалонаткиназы среди голландцев составляет около 1:65. Кроме того, случаи HIDS были зарегистрированы и среди жителей других европейских странах – Англии, Германии, Италии, Чехии, Турции, а также в США и Японии. Соотношение пациентов мужского и женского пола составляет 3:2. У большинства пациентов первые симптомы заболевания появляются на первом году жизни (средний возраст – 6 мес.). Приступы лихорадки сохраняются в течение всей жизни, хотя после подросткового возраста может отмечаться сокращение частоты и интенсивности приступов.
Клинически гипер IgD-синдром проявляется повторными приступами лихорадки, которые возникают каждые 4–8 недель и длятся 3–7 дней, хотя временные интервалы у разных пациентов могут отличаться.
Приступы манифестируют высокой, пикообразной лихорадкой, которой предшествует озноб у 76% пациентов. В течение приступов у 72% пациентов отмечаются боли в животе, у 56% – рвота, у 82% – диарея, у 52% – головная боль. Суставной синдром также достаточно характерен для воспалительных атак при HIDS: в 80% случаев имеют место моноартралгии, в 68% – недеструктивный артрит. У 94% пациентов отмечается увеличение периферических лимфатических узлов, нередко обнаруживается спленомегалия. У около 82% пациентов во время части приступов появляются высыпания на коже – чаще всего, эритематозные пятна и папулы, иногда – петехии и пурпура. Нередко выявляется афтозный стоматит и изъязвление слизистой оболочки наружных половых органов и влагалища. Серозиты – редкое проявление приступов при HIDS. Несмотря на частые воспалительные атаки, ни у одного из пациентов с гипер IgD-синдромом не развивается амилоидоз.
К факторам, которые могут провоцировать приступ, относятся прививки, травмы, хирургические вмешательства, физическое и эмоциональное напряжение.
Для пациентов с HIDS типично выявление постоянно повышенного сывороточного уровня IgD – более 100 ед./мл, хотя у части больных уровень IgD может быть нормальным. Уровень IgD не коррелирует ни с тяжестью течения заболевания, ни с уровнем активности мевалонаткиназы, ни с генотипом. У примерно 82% пациентов определяют также повышенный уровень сывороточного IgA.
В период приступа повышаются уровни острофазовых показателей и концентрация мевалоновой кислоты в моче.
Диагностические критерии гиперIgD-синдрома (Simon A. et al., 2001):
- Повышение уровня IgD в сыворотке крови (более 100 ед./мл) в 2 пробах, взятых с интервалом не менее 1 месяца.
- В период приступов:
- повышение СОЭ и лейкоцитоз;
- острое начало лихорадки (температура тела выше 38,5°C);
- повышение уровня сывороточного IgA;
- увеличение шейных лимфоузлов;
- абдоминальные симптомы (рвота, диарея, боль в животе);
- кожные проявления (эритематозные пятна и папулы);
- артралгии и/или артрит;
- спленомегалия.
- Рецидивирующий характер приступов.
Гипер IgD-синдром нужно дифференцировать с мевалоновой ацидурией, при которой помимо приступов лихорадки отмечается задержка ПМР и физического развития, атаксия, катаракта и черепно-лицевой дизморфизм (микроцефалия, треугольное лицо, гипоплазия крыльев носа). У детей с мевалоновой ацидурией активность мевалонаткиназы не обнаруживается, и пациенты обычно погибают в раннем возрасте, тогда как при HIDS активность фермента составляет около 1–7% от нормы, и прогноз для жизни благоприятный.
В лечении приступов лихорадки при гипер IgD-синдроме колхицин и стероиды неэффективны. Недавние исследования показали, что применение симвастатина (ингибитора 3-гидрокси-3-метилглутарил-коэнзим редуктазы, которая предшествует мевалонаткиназе) в дозе 80 мг/сут. способствовало уменьшению экскреции мевалоновой кислоты и сокращению длительности приступов лихорадки.
Продолжение в следующем номере.
ДетальнішеГЕМАНГІОМИ У ДІТЕЙ
Гемангіома – це доброякісна пухлина судинного ендотелію. Відноситься до групи вродженої патології судин, внаслідок дефекту їх ембріонального розвитку.
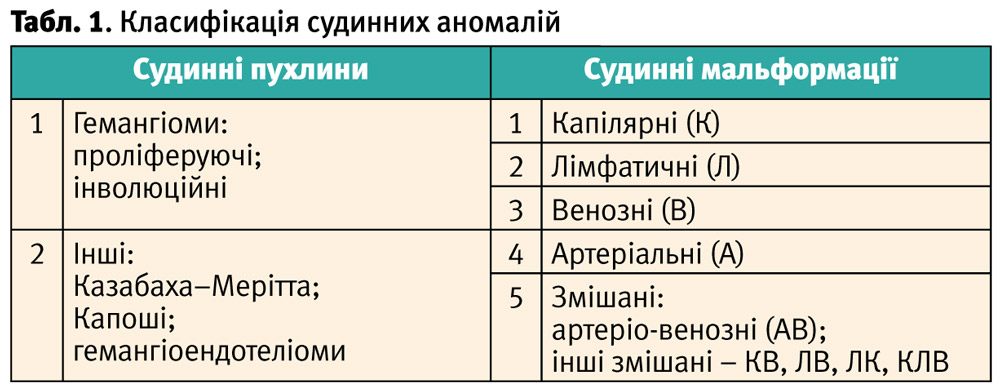
Згідно з класифікацією, запропонованою Міжнародним товариством досліджень судинних аномалій (ISSVA), усі судинні аномалії поділяються на дві основні групи:
- Судинні пухлини.
- Судинні мальформації (таблиця 1).
Основною різницею між цими групами судинних аномалій є те, що вони мають різну етіологію виникнення, різний перебіг свого розвитку, відмінну гістологічну будову та клітинні маркери.
Причини виникнення гемангіом чітко не встановлені. Ці судинні пухлини є наслідком гіперплазії ендотелію судин. Зустрічаються гемангіоми, за даними різних авторів, приблизно у 10% всіх новонароджених, частіше у дівчаток, ніж у хлопчиків, у співвідношенні 6:1. При цьому, частіше у недоношених дітей та дітей з низькою масою тіла. Лише у 30% випадків гемангіома є помітною відразу після народження, у решти дітей проявляється у перші тижні життя. Найчастіша локалізація – голова, шия, верхня половина тіла (60%) та кінцівки (15%).
Гемангіоми характеризуються стадійним перебігом. Виділяють фазу проліферації та фазу інволюції. Проліферативна фаза характеризується прогресивним ростом пухлини, яка добре васкуляризується харчуючими артеріями, триває переважно до 7–8-місячного віку і має два періоди інтенсивного росту: 3–4 місяці та 6–8 місяців. Фаза інволюції триває до 3–7 років.
До 7-річного віку 70–80% гемангіом регресує. Регрес гемангіоми може завершитись:
- повним її зникненням;
- депігментацією шкіри на місці гемангіоми;
- сплощенням, атрофією тканин;
- надлишком поморщеної шкіри з помаранчевим відтінком;
- рубцюванням.
Класифікація гемангіом
Найбільш поширеною класифікацією гемангіом є така:
- Прості або капілярні.
- Кавернозні.
- Змішані.
- Комбіновані.
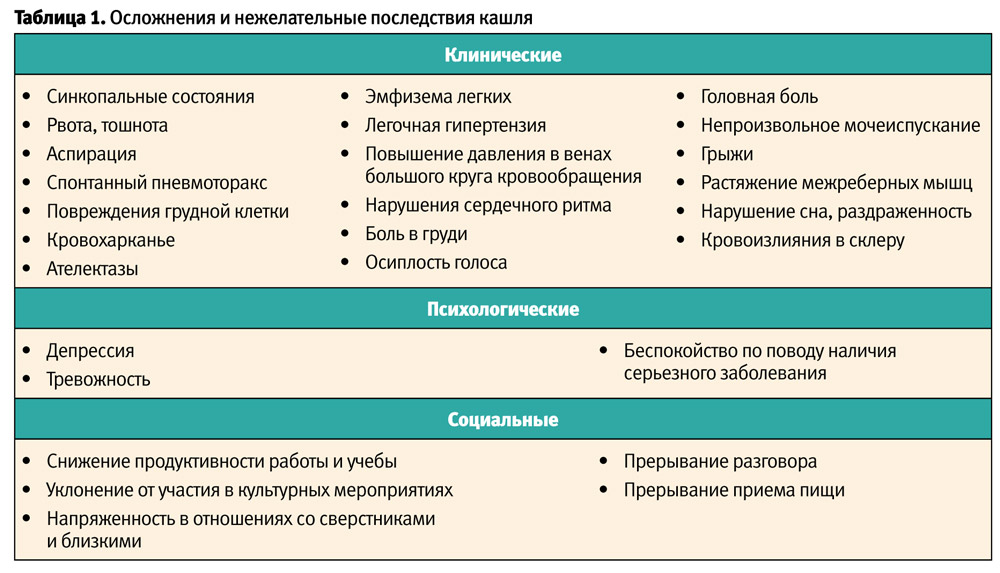
Капілярна гемангіома клінічно проявляється утворенням яскраво-червоного кольору з характерною будовою, спочатку у вигляді плями, яка з ростом гемангіоми підноситься над поверхнею шкіри та нагадує за будовою малину. При натисканні на пухлину вона блідне, але швидко відновлює колір при припиненні компресії (фото 1).
Кавернозна гемангіома розташована в основному в підшкірній клітковині, має вузлувату будову і на шкірі проявляється у вигляді синюшного забарвлення у місці її локалізації. При пальпації відчувається її вузлуватість. При компресії спостерігається виражений «симптом губки» (фото 2).
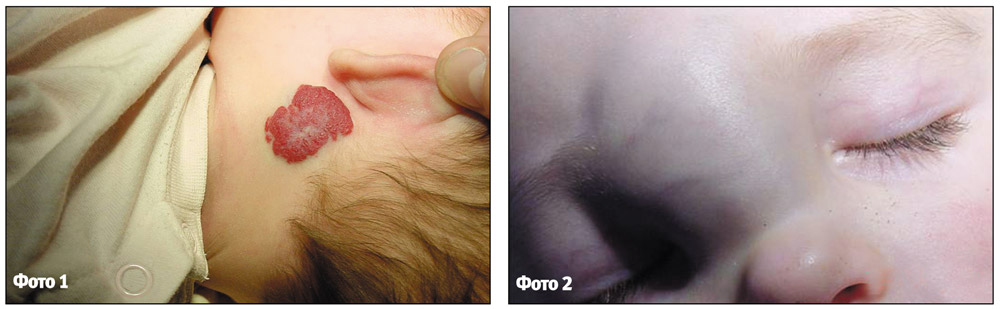
Комбінована гемангіома представляє собою поєднання поверхневої та підшкірної гемангіом, капілярної та кавернозної (фото 3).
Змішана гемангіома складається з пухлинних клітин, що виходять з ендотелію судин та клітин інших тканин. Зовнішній вигляд, колір та консистенція пухлини визначаються складом її тканини (фото 4).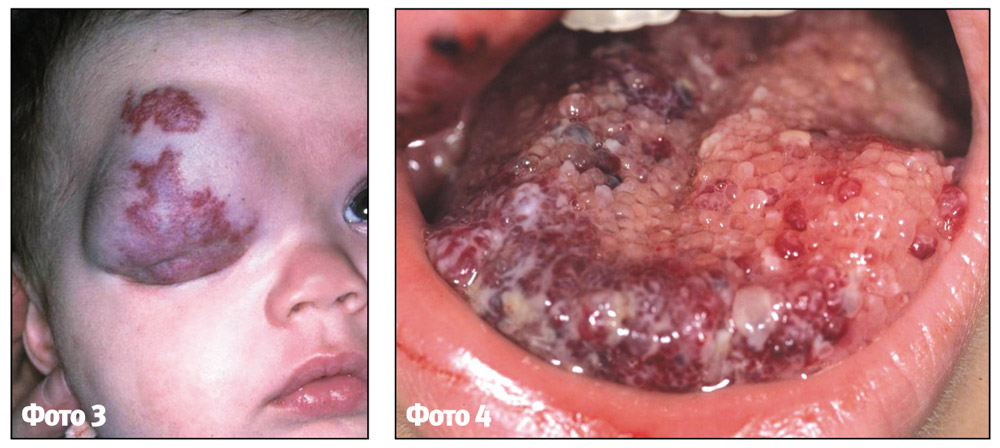
Гемангіому відносять до доброякісних пухлин. Вона переважно має капсулу, але може рости дифузно та проростати у підшкірно-жирову клітковину й глибше розташовані тканини та органи. У літературі описані випадки локалізації гемангіом у тканині головного та спинного мозку, на слизових ротоглотки, дихальних шляхів та шлунково-кишкового тракту, у паренхіматозних органах, з яких найчастіше уражається печінка.
Відповідно до медичних протоколів, дитині з множинними гемангіомами шкіри обов’язково слід виконати нейросонографію та ультразвукове обстеження внутрішніх органів. Множинні гемангіоми та гемангіоми, розмір яких перевищує 5 см у діаметрі, можуть бути причиною злоякісної тромбоцитопенії (синдром Казабаха–Мерітта) та гіпотиреоїдизму.
У фазі проліферації гемангіома прогресивно збільшується у розмірах та змінює колір на яскраво-червоний. Інтенсивний ендотеліальний ріст в проліферативних гемангіомах стимулюється ангіогенним факторами. Вони безпосередньо діють на ендотелій судин, стимулюючи мітоз, або через хелперні клітини (тучні клітини, макрофаги). Тучні клітини вивільняють гепарин, який стимулює ангіогенез, міграцію ендотеліальних клітин та ріст капілярів. Ангіогенна дія гепарину може блокуватися протаміном. Крім протаміну ангіогенез гальмується речовинами, які входять до складу глюкокортикостероїдів, що підтверджується їх позитивною дією на інволюцію гемангіом.
Діагностика
 Діагностика гемангіом у переважній більшості випадків не є важкою. У 90% випадків діагноз лікар ставить на основі анамнезу та огляду дитини. Якщо після первинного огляду діагноз залишається не з’ясованим, тоді застосовуються додаткові методи обстеження:
Діагностика гемангіом у переважній більшості випадків не є важкою. У 90% випадків діагноз лікар ставить на основі анамнезу та огляду дитини. Якщо після первинного огляду діагноз залишається не з’ясованим, тоді застосовуються додаткові методи обстеження:
- Клінічний – повторні огляди та спостереження за динамікою процесу.
- Ультразвукове обстеження.
- Магнітно-ядерна томографія.
- Ангіографія.
- Тепловізорне обстеження.
- Комп’ютерна томографія (при підозрі на ураження кісток).
- Визначення кількісних та якісних показників ангіогенних факторів.
Ускладнення
Найпоширенішими ускладеннями, які можуть виникати в процесі розвитку гемангіоми є:
- кровотечі зовнішні та внутрішні);
- виразкування;
- інфікування (навіть сепсис);
- некроз пухлини;
- тромбоцитопенія споживання (синдром Казабаха–Мерітта), ДВЗ-синдром;
- гостра серцева недостатність (при гемангіоматозі печінки);
- косметичні та функціональні ускладнення (втрата зору, слуху, деформації з порушенням функції органів).
Диференціальна діагностика
Диференціальну діагностику гемангіом найважче проводити з судинними мальформаціями, які візуально часто нагадують гемангіому. Найбільш характерними відмінностями судинних мальформацій є наступні:
а) судинні мальформації не мають стадійності розвитку, вони повільно прогресивно розвиваються та збільшуються пропорційно до росту дитини;
б) частіше уражають кінцівки та тулуб;
в) в переважній більшості є змішаними пухлинами у вигляді артеріо-венозних, лімфо-венозних та артеріо-лімфовенозних утворів;
г) часто є проявами синдромів (Мафуччі, Кліппеля–Треноне, Вебера).
Лікування
Лікування гемангіом інколи є складним і вимагає, в деяких випадках, спільних дій цілої команди спеціалістів:
- хірургів;
- дерматологів;
- сонографістів;
- радіологів;
- косметологів;
- генетиків.
У лікуванні гемангіом застосовують різні методи. Вибір того чи іншого методу лікування залежить від багатьох факторів:
1) типу гемангіоми;
2) розмірів;
3) локалізації;
4) інтенсивності росту;
5) досвіду лікаря;
6) можливостей лікувальної установи.
Методи лікування гемангіом:
- Хірургічний. Найчастіше використовується при відсутності повної інволюції пухлини та, коли не досягнуто бажаного ефекту іншими методами лікування. Широко застосовується при кавернозних гемангіомах. Досягнення косметичної хірургії розширили можливості хірургічного лікування гемангіом з хорошим косметичним ефектом.
- Лазеротерапія. Цей метод широко застосовується при капілярних гемангіомах у вигляді «винних плям». Вимагає відповідного оснащення.
- Стероїдотерапія. Широко застосовується з хорошими результатами, але необхідно пам’ятати про побічну дію стероїдів на організм дитини, що обмежує можливості цього методу.
- Емболізація судин. Використовується в окремих випадках за строго визначеними показами з метою збіднення артеріального кровопостачання пухлини, що сприяє її інволюції.
- Електрокоагуляція. Можна використовувати при павучкових ангіомах або, якщо розміри гемангіоми не досягають 1 мм.
- Кріотерапія та радіотерапія практично не застосовуються.
- Лікування пропранололом. Пропранолол – неселективний бета-адрено-блокатор. Лікувальна дія пропранололу при гемангіомах вивчена не так давно. Але за останні роки цей метод лікування вважається найперспективнішим і дає найкращі результати. В Україні широко використовується аналог пропранололу – анаприлін. Дія пропранололу на гемангіому проявляється гальмуванням ангіогенезу, сповільненням тубулогенезу ендотеліальних клітин та збідненням кровопостачання пухлини за рахунок спазму судин. Дія пропранололу на гемангіому проявляється найкраще у фазу проліферації пухлини, тому згідно рекомендацій препарат застосовують у дітей віком до одного року.
Результати лікування гемангіом залежать від своєчасного їх виявлення та своєчасного скерування дитини до спеціаліста медичного центру, у якому доступні сучасні методи діагностики та лікування цієї патології.
ДетальнішеОСОБЛИВОСТІ ПСИХОЛОГІЧНОГО ПРИЙНЯТТЯ БАТЬКАМИ ДІТЕЙ З ОСОБЛИВИМИ ПОТРЕБАМИ
Серед напрямків психологічної реабілітації у Сумському обласному центрі соціальної реабілітації дітей-інвалідів особливе місце відводиться психологічній підтримці сімей, які виховують дітей-інвалідів. Основними засобами, якими користується психолог у своїй діяльності під час роботи з батьками, є: діагностика, психокорекція, консультація, психологічне просвітництво
Особлива увага приділяється створенню сприятливого психоемоційного клімату (налагодженню батьківсько-дитячих взаємовідносин, корекції неадекватних реакцій поведінки та емоційних реакцій батьків, які мають дитину-інваліда; формуванню навичок адекватного спілкування з навколишнім світом). Адже, саме в сім'ї відбуваються процеси, від яких значною мірою залежить доля дитини-інваліда: формується самооцінка, визначається соціальна роль і позиція. Сім'я, як реабілітаційне середовище, не тільки виявляє турботу, надає фізичну допомогу, психологічну підтримку, допомагає адаптуватись до сучасного способу життя, а й організовує відновлююче лікування, навчання, допомагає обрати професію. Міжособистісні стосунки в сім'ї, ставлення батьків, родичів до дитини-інваліда, обладнання місця проживання, навчання побутовим навичкам – все це фактори, які позитивно чи негативно відбиваються на реабілітації дитини з особливими потребами.
Родини наших вихованців завжди у центрі уваги, адже батьки найбільш зацікавлені у вирішенні проблем своєї дитини. Сім’ї, у яких виховуються діти з відхиленнями у розвитку, живуть під тягарем багаточисельних труднощів, не кожний батько чи мати виявляються здатними прийняти недугу дитини, адекватно реагувати на проблеми, що постійно виникають. Відомо, що психотравмуюча ситуація, а також колосальні емоційні навантаження на членів сім'ї, у зв'язку з довготривалим стресом, чинять негативний вплив на психіку батьків та ускладнють їхнє ставлення до дитини. Багато батьків у ситуації, що склалася, виявляються безпорадними. Їхній стан можна охарактеризувати як внутрішній (психологічний) і зовнішній (соціальний) глухий кут. Декого трагічність ситуації навіть ламає. А саме особистісні якості батька та матері визначають можливості соціалізації дітей та адаптації до життя, тобто їхнє майбутнє.
Для того, щоб допомогти батькам дитини з вадами розвитку, важливо спробувати зрозуміти, що відбувається з людиною, коли її дитині встановлюють інвалідність, як це впливає на життя.
Простежуючи переживання батьками трагедії народження неповносправної дитини, різні дослідники (Райт, Дуккан, Дрокар, Шухард) дійшли висновку про закономірну зміну їхніх емоційних станів на шляху до адаптації. Ці стани характерні для переживання будь-якого горя, адже народження дитини з функціональними обмеженнями є для батьків тим поштовхом, який запускає в організмі процеси «переживання горя».
Детально опис переживань, як процес адаптації до нової життєвої ситуації, наводиться у монографії Е. Шухарда. Автор пропонує свою періодизацію кризових станів емоційної сфери батьків:
1) шок, невизначеність (стан панічного жаху перед невідомим, відчуття того, що руйнується звичне «нормальне» життя);
2) поступове розуміння, визначеність (протиріччя між розумінням проблеми на раціональному рівні та її неприйняття на рівні емоцій та почуттів);
3) агресія (прояви негативних почуттів у вигляді емоційних спалахів, у результаті чого виникає агресія, спрямована на оточуючих);
4) заперечення – віра у зцілення, помилковість діагнозу (період тривалого і виснажливого мандрування від лікаря до лікаря, а далі – до знахарів, екстрасенсів із надією знайти чудо, яке зробить дитину здоровою);
5) депресія (переживання почуття безнадійності, апатії та відчаю у зв'язку з безуспішністю пошуку способів зцілення дитини);
6) прийняття факту порушення розвитку (поява нового сенсу життя);
7) активізація (поява інтересу до навколишнього життя, відкриття нових можливостей самореалізації);
8) солідарність (об’єднання з іншими батьками, які мають аналогічні труднощі).
Ставлення батьків до дітей з порушенням психофізичного розвитку відіграє першорядну роль. Тому вкрай важливе значення має такий фактор, як психологічні особливості прийняття батьками дітей з особливими потребами.
У практику дитячо-батьківських стосунків термін «прийняття» увійшов завдяки представнику гуманістичної психології К. Роджерсу, який трактував прийняття як безумовно позитивне ставлення до дитини незалежно від того, чи радує вона дорослих у даний момент. Водночас важливо зрозуміти: безумовне прийняття не означає буквально, що інші значущі люди повинні пробачати або схвалювати усе, що дитина робить або говорить. Насправді йдеться про створення такого сімейного оточення, у якому малюка хвалять і визнають як повноцінного члена родини, котрий інколи може бути нестерпним, але якого, все ж таки, люблять. Тобто, виховання дітей з безумовним прийняттям та позитивною увагою забезпечує надійну основу для їхнього становлення та повноцінного функціонування у дорослому житті. Стосовно системного підходу до типологізації прийняття дитини і батьківського ставлення готовність батьків до повного прийняття дитини визначається такими кроками: дозволом їй бути такою, якою вона є; увагою до її почуттів і думок, умінням почути та зрозуміти їх; здатністю підтримати дитину, усвідомлюючи, що її цінності та системи поглядів можуть не збігатися з батьківськими; повагою до позиції дитини, вірою в її сили та можливості; готовністю ділитися власними цінностями і поглядами, створюючи тим самим можливість дитині розуміти інших. За усім цим стоїть безкорислива, істинна любов батьків до дітей, яка допомагає дорослим відмовитися від фіксації на слабкостях, недоліках, недосконалостях, спрямовує виховні зусилля на підкріплення позитивних якостей особистості дитини, на підтримку її сильних сторін. Адже саме у такій атмосфері, де безоцінно приймаються будь-які індивідуальні відмінності, де любов і прихильність виражаються відкрито, де помилки допомагають здобути новий досвід, де спілкування відкрите й довірливе, де особиста відповідальність і чесність – обов'язкові складові взаємин, може сформуватися почуття самоцінності у кожного члена сім'ї.
Психологічне прийняття – це особлива позиція щодо себе та своєї дитини, яка має когнітивний, емоційно-смисловий та поведінковий компоненти. Когнітивний (пізнавальний) компонент виявляється у тому, що батьки мають необхідні знання про дитину, причому не лише зовнішні, але й інтуїтивні – вміння визначати потреби дитини. Емоційно-смисловий компонент виражається у повному прийнятті батьками дитини з особливими потребами як самостійної цінності: дитина для батьків не є засобом самореалізації. Поведінковий компонент характеризується компетентною взаємодією батьків з дитиною, адекватною до потреб дитини, здатною до відповідального вибору.
Особливості прийняття батьками дітей з вадами розвитку можна поділити на:
- адекватне – прийняття дитини «такою, як вона є». Батькам важливо, що дитина є, і що саме вона – їхня дитина, незалежно від того, яка вона і що вміє робити. Така безумовна любов допомагає батькам правильно оцінити, до чого привели їх виховні зусилля і підказує, у якому напрямку слід рухатись далі.
- неадекватне — це «відторгнення» дитини, негативне ставлення до неї. У батьків це ставлення може проявлятись у роздратуванні по відношенню до дитини, в ігноруванні її інтересів, в силу їх примітивності, а також у бажанні покарати дитину.
- суб’єктивне – дитина з особливими потребами сприймається як суб’єкт соціальних відносин, тобто як повноправний член суспільства. Ані батьки, ані найближче оточення не фокусуються на інвалідності дитини, що є сприятливим фактором для її подальшої адаптації у суспільстві, а також сприяє толерантному ставленню до людей з обмеженими можливостями у суспільстві.
- об’єктивне – дитина сприймається як особистість, яка віддалена від суспільства. У батьків воно проявляється у бажанні «сховати» дитину від усіх, що в свою чергу призводить до неадекватної форми взаємодії дитини з соціальним середовищем, і виникненням комунікативних проблем та перешкод. Через це відносини з людьми у свідомості дитини фарбуються у несприятливі тони, для неї характерні відчуженість, тривожність, агресія.
Таким чином, під психологічним прийняттям ми розуміємо цілісну систему різноманітних почуттів до дитини, поведінкових стереотипів, що практикуються по відношенню до неї, особливостей сприйняття та розуміння характеру дитини, її вчинків. Це визнання права дитини на властиву їй індивідуальність, несхожість на інших, у тому числі несхожість на батьків. У ситуації з дитиною з особливими потребами — це найбільш актуально. Також у рамках особистісного прийняття або неприйняття дитини формується психологічний та емоційний стан батьків і рівень спілкування у родині, коли з'являється дитина з особливими потребами.
Особистісне прийняття або неприйняття батьками дефекту дитини також лежить в основі виховного впливу батьків і впливає на визначення батьками моделі виховання та відіграє першочергову роль у подальшому становленні таких дітей у суспільстві.
Рекомендації батькам, які мають дітей-інвалідів.
Прийміть ситуацію як даність, змиріться з нею, не думайте про те, як і чому це трапилось, розмірковуйте про те, як з цим жити далі. Пам’ятайте, що всі ваші страхи та «чорні думки» дитина відчуває на інтуїтивному рівні. Тому, якщо ви не хочете, щоб ваша дитина росла нервовою, похмурою, постарайтесь знайти в собі сили з оптимізмом дивитись в майбутнє.
Ніколи не жалійте дитину через те, що вона не така, як усі.
Даруйте дитині свою любов та увагу, але не забувайте, що у всіх членів родини була можливість саморозвитку та повноцінного життя.
Організовуйте свій побут так, щоб ніхто в родині не відчував себе «жертвою», відмовляючись від власного особистого життя. Не захищайте дитину від обов’язків та проблем. Якщо стан дитини дозволяє, вигадайте їй прості домашні обов’язки, намагайтеся навчити дитину піклуватись про інших. Вирішуйте усі справи разом з нею.
Надайте дитині самостійність у діях та прийнятті рішень. Стимулюйте її пристосовану активність; допомагайте у пошуку своїх прихованих можливостей. Розвивайте вміння та навички самообслуговування.
Слідкуйте за своєю зовнішністю та поведінкою. Дитина повинна пишатися вами. Не бійтесь відмовити дитині в чому-небудь, якщо вважаєте її вимоги надмірними. Однак, проаналізуйте кількість заборон, з якими стикається ваша дитина. Продумайте, чи всі вони обґрунтовані, чи не має можливості скоротити обмеження, зайвий раз проконсультуйтесь з лікарем чи психологом.
Частіше розмовляйте з дитиною. Пам’ятайте, що ні телевізор, ні радіо не замінять вас.
Не обмежуйте дитину у спілкуванні з ровесниками.
Не відмовляйтесь від зустрічі з друзями, запрошуйте їх у гості. Нехай у вашому житті знайдеться місце і високим почуттям, і маленьким радостям.
Частіше користуйтесь порадами педагогів та психологів. Кожне певне захворювання дитини-інваліда потребує специфічного догляду, а також спеціальних знань та умінь. Більше читайте, і не лише спеціалізовану літературу, але й художню.
Спілкуйтесь з родинами, де є діти-інваліди. Є важливо не лише для вас, але й для дитини, якій ви можете зробити послугу на все життя, знайшовши для неї друзів або (що дуже часто буває) супутника життя.
Не докоряйте собі. В цьому випадку велика імовірність того, що дитина виросте психологічним монстром, а це неминуче посилить її соціальну дезадаптацію та збільшить страждання. У тому, що у вас хвора дитина, ви не винні.
Детальніше
НОВАЯ НДЕЖДА ДЛЯ ДЕТЕЙ С АУТИЗМОМ
«Раннее выявление детей с аутизмом очень важно, так как рано начатая коррекция коммуникативных нарушений дает возможность получить значительную положительную динамику в психическом состоянии ребенка. «Учебное Пособие по Аутизму» в очень доступной форме, наглядно, лаконично подает самую необходимую информацию по выявлению аутизма у детей раннего возраста. Книга прекрасно иллюстрирована. Думаю, что такая книга будет необходима не только педиатрам и семейным врачам, но и другим специалистам, родителям, которые также найдут в ней полезную информацию».
Людмила Литвин, детский психиатр,
Президент Всеукраинской ассоциации детских психиатров.

20 ноября в Киеве состоялся совместный пресс-ланч Фонда «Дитина з майбутнім» и Украинской Медицинской Миссии, в рамках которого было презентовано первое учебное пособие по аутизму для педиатров, родителей и всех специалистов, работающих в этой области. «Учебное Пособие по Аутизму» было составлено Линдой Ли, исполнительным директором ОПАС, на основе работ ведущих медицинских экспертов и педагогов.Во время пресс-ланча спикеры обсуждали самый актуальный вопрос – несвоевременность постановки диагноза, которая связана с незнанием настоящих симптомов аутизма в среде украинских педиатров и профильных специалистов.
Новое пособие, которое должно заполнить существующий пробел, будет распространяться по всем детским поликлиникам Украины. О необходимости появления такого пособия говорили давно, ведь проблема с диагностикой аутизма начинается как раз на уровне педиатров. Зачастую специалисты первичного уровня не видят у ребенка существующую проблему и списывают все на особенности характера, предлагая родителям занять выжидательную позицию, тем самым упуская драгоценное время. Использование методик, представленных в пособии, позволит украинским педиатрам своевременно направлять детей к узкопрофильным специалистам для постановки окончательного диагноза.
Говоря о важности своевременной диагностики, Консул международной ассоциации «Аутизм Европа», учредитель благотворительного фонда «Дитина з майбутнім» Инна Сергиенко отметила: «Почему мы боремся за раннюю диагностику? Чтобы как можно раньше начать реабилитацию ребенка. Чем раньше с ним начнут работать специалисты, тем больше можно помочь. В таком случае 60–70% детей с аутизмом выходят на приемлемый уровень социализации. Поэтому отдельное спасибо Алене Васильевне Терещенко, начальнику Управления материнства, детства и санаторного обеспечения МОЗ Украины, которая помогает нам в распространении информации по поликлиникам Украины».
По словам Инны Сергиенко, министерство здравоохранения поможет дойти Пособию до каждой детской поликлиники и таким образом помочь в ранней диагностике аутизма.
Выступивший на пресс-ланче детский врач и президент Медицинской Миссии в Украине Джеймс Пипон, рассказал о том, как появилась идея издания Пособия: «Во время тренингов для специалистов мы увидели, что в Украине существует проблема ранней диагностики аутизма, поэтому решили перевести наше пособие на русский язык. На сегодняшний день мы помогли издать 10 тысяч таких пособий для распространения в Украине».
Что касается адаптации и распространения Пособия, то этим в Украине занимается Фонд помощи детям с синдромом аутизма «Дитина з майбутнім». Директор фонда Лариса Рыбченко в своем выступлении говорила о необходимости переаттестации специалистов, от которых зависит своевременное выявление детей с аутизмом: «Сегодня у нас не более десятка детских психиатров, которые действительно могут правильно диагностировать аутизм. Поэтому мы будем требовать от Министерства здравоохранения провести переаттестацию всех психиатров и педиатров».
Скачать пособие можно по адресу: http://cwf.com.ua/ru/documents/830-uchebnoe-posobie-po-autizmu
Детальніше