

Современные возможности диагностики открыли доступ к высокоразрешающим методам анализа, позволяющим идентифицировать тонкие генетические аномалии на этапе преимплантационной и пренатальной диагностики. В свете этого, важную роль приобретает задача поиска ассоциаций микроструктурных хромосомных аномалий с различными состояниями плода, причины возникновения которых до недавнего времени не удавалось обнаружить. Расширение возможностей также ставит задачу пересмотра алгоритмов ведения пациентов, ведь при подозрении на наличие аномалии ограничение исследования пренатальным кариотипированием может быть необоснованным.
Известно, что естественный отбор является основным фактором, регулирующим процессы эволюции на различных уровнях иерархии генетических систем, элиминируя сегрегационный и мутационный груз (Тимченко О. И. и др., 2015), который в медицинской практике мы воспринимаем в виде бесплодия и репродуктивных потерь.
Однако, при совместимых с жизнью аномалиях, вероятность самопроизвольного прерывания беременности снижена. С этой точки зрения микроделеционные синдромы являются наиболее коварными. Зачастую, они не имеют специфических клинических проявлений, а степень их выраженности определяется размером делетированного участка и вовлеченными генами. Дети, рожденные с микроструктурной хромосомной аномалией, могут иметь различной степени выраженности задержку психофизического развития, врожденные аномалии, которые будут сопровождаться социальной дезадаптацией и инвалидностью ребенка (Nevado J et. al., 2014:210-214; Watson C. T. et al, 2014: 215-244).
Клиническое наблюдение
Беременная К, 1979 г. р. (на момент обращения – 36 лет), обратилась в Клинику репродуктивной медицины «Надия» для проведения пренатального скрининга первого триместра. Вес 74 кг, рост 173 см. ИМТ 24,7.
Беременность вторая, природная, одноплодная. Срок 12 нед. 2 дня. В анамнезе одна внематочная беременность.
Семейный анамнез со слов супругов, не отягощен. Пациентка отмечала склонность к возникновению аллергических реакций. Вредные привычки отрицает. Группа крови О(I), RhD-фактор: положительный. Супруги в родстве не пребывают.
Результаты УЗ-исследования:
ЧСС плода 174 уд./мин., КТР 58,4 мм, ТВП 4,8 мм, МТР 19 мм, ОГ 67,5 мм, ОЖ 53,5 мм, длина бедренной кости 7,3 мм. Плод выглядит обычно.
Сопутствующий диагноз: множественная миома матки.
Результаты биохим. исследований:
Свободный b-ХГЧ – 12,6 МЕ/л (0,312 МОМ)
РАРР-А – 0,356 МЕ/л (0,201 МОМ).
Пациентке было рекомендовано кариотипирование плода путем инвазивной процедуры, от которого она воздержалась в пользу неинвазивного пренатального тестирования хромосомных аномалий плода по крови беременной (НИПТ) Prena Test (провайдер Life Code xx, GE / Клиника «Надия»). Для проведения тестирования пациентка обратилась в срок 15 недель беременности.
Согласно результатам НИПТ, плод пациентки К. был мужского пола и не являлся носителем анеуплоидий по 13, 18, 21 и половым хромосомам (z-score Ch13 0.4 (REF<3.9), Ch18 1.2 (REF<3.2), Ch21 2.0 (REF<3.0), ChX -3.1 (REF<-3.0), ChY 8.6 (REF>3.0).
Пациентке были разъяснены ограничения исследования НИПТ, в частности относительно микроделеционных синдромов, сбалансированных хромосомных перестроек, аномалий хромосом, не вовлеченных в исследование, а также низкоуровневого мозаицизма.
Со значительной задержкой от даты рекомендации врачом инвазивной процедуры пациентка прошла процедуру амниоцентеза с целью кариотипирования плода в срок беременности 17 нед. 2 дня, но без последующего обращения к генетику для разъяснения результатов кариотипиования.
Результат кариотипирования: 46,XY. Нормальный мужской кариотип амниоцитов (тип окраски GTG, разрешение 400–450 бэндов на диплоидный набор).
Цитологическое исследование амниотический жидкости: в препаратах эпителий 5–10 в п. з., лейкоциты 0–5 в п. з., эритроциты 5–10 в п. з. не изменены.
УЗ-исследование плода в сроке 20 нед. 6 дней: МТР 43,3 мм, ЛЗР 57,3 мм, ОГ 158 мм, ОЖ 139,2 мм. Сердцебиение, движение плода визуализируется. Детальная оценка строения тела плода: без особенностей, аномалии развития не визуализируются. Признаки задержки развития плода. Краевое предлежание плаценты, степень зрелости 0 по Grannum. Множественная миома матки.
После дополнительного УЗИ в сроке беременности 22 нед. 2 дня пациентка принимает настоятельную рекомендацию медико-генетического консультирования, на которое она обращается в сроке беременности 22 нед. 6 дней, понимая, что результат консультирования никоим образом не повлияет на вынашивание беременности (согласно законодательству Украины).
При проведении консультирования установлено, что родной брат пациентки родился с пороком кишечника и почек, умер в неонатальном периоде.
Со стороны супруга в предыдущем браке ребенок умер от неоперабельного порока сердца. Пациентке был разъяснен риск наличия микроструктурных хромосомных аномалий (~5,5%). Для определения оптимальной стратегии сопровождения новорожденного, учитывая результаты медико-генетического консультирования, пациентке предложена диагностика методом сравнительной геномной гибридизации.
Пациентке еще раз было разъяснено, что любой результат проведенного исследования уже не сможет изменить дальнейшую тактику ведения беременности или изменить ее исход. Пациентка еще была ранее осведомлена о том, что прерывание беременности законодательно разрешено до 22 недель.
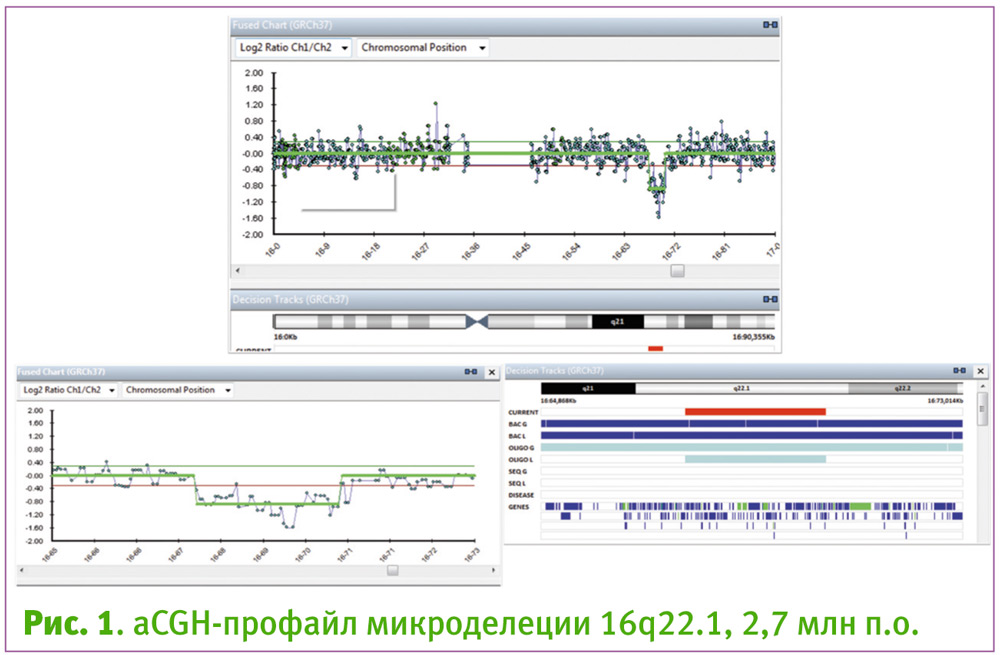
Учитывая настоятельное желание женщины максимально точно определить прогноз здоровья ребенка и выбрать дальнейшую тактику сопровождения новорожденного, для проведения чип-сравнительной геномной гибридизации была использована криоконсервированная культура амниоцитов. 1 мкг выделенной ДНК (Nucleo Spin Blood, Macherey-Nagel, GE) гибридизован на чипах Illumina Cyto Chip ISCA 44kv.2.0 (USA). Выявлена интерстициальная микроделеция хромосомы 16 в пределах бэнда 16q22.1 с координатами 67,654,596-70,383,861 и протяженностью 2,7 млн п. н. Срок беременности на момент заключения – 24 нед. 1 день.
Результирующий кариотип: 46,XY.arr 16q22.1 (67,654,596-70,383,861) x1.
Согласно международным базам данных, клиническое значение делеции данного участка окончательно не установлено. При этом известно, что делеция перекрывает вероятно патогенный регион на 38%, а 16% самой делеции являются нормальным полиморфизмом по числу копий (CNV). На основании приведенной информации сделано заключение, что делеция имеет возможно патогенное значение.
Локализированные в зоне делеции гены и ассоциированная с ними патология приведены в табл. 1.
При наличии данной делеции отмечен риск задержки психомоторного развития, поведенческих отклонений, органических нарушений головного мозга на уровне 63%. Учитывая полученные результаты, рекомендована консультация генетика и детского невролога после рождения ребенка, ДНК-диагностика микроструктурных хромосомных аномалий супружеской паре с целью исключения наследования делеции и определения ее возможного клинического значения.
Роды. Ребенок от ІІ беременности, протекавшей на фоне преэклампсии средней степени, предлежания плаценты, плацентарной дисфункции, родился путем ургентного кесарева сечения (дистресс плода) на 31–32 неделе гестации, пятикратное обвитие пуповины вокруг шеи. Оценка по Апгар 3–4 балла. Масса 1190 г, длина 39 см, окружность головы 26 см, груди – 24 см. Диагноз первичный: дыхательные нарушения легкой степени тяжести. Стигмы: микроцефалия, микрогнатия, нарушение пальцевого ряда на ногах, арахнодактилия. Недоношенность. Малый к сроку гестации. Первые 3 недели жизни ребенок находился без дополнительной дыхательной поддержки.
На 22 сутки переведен на искусственную вентиляцию легких (ИВЛ) на 9 дней ввиду развившейся пневмонии.
В 1 мес. 16 дней диагностирован врожденный порок сердца: функционально значимый открытый артериальный проток (до 3 мм). Двухстворчатый аортальный клапан. Открытое овальное окно. Проведено хирургическое закрытие артериального протока.

В возрасте 3 недель диагностирована ретинопатия недоношеных 1–2 ст.
В начале второго месяца жизни появилась паховомошоночная правосторонняя грыжа. Сохранен двусторонний крипторхизм.
В течение второго месяца жизни ребенок трижды повторно переводился на ИВЛ ввиду затяжных апноэ (видимо, центрального генеза), не поддающихся медикаментозной коррекции.
В начале третьего месяца развилась тяжелая форма бронхолегочной дисплазии, ребенок на вентиляции. Смерть наступила на 77 сутки жизни.
Продолжительность жизни – 77 суток, из них ИВЛ – 33 суток.
У ребенка не выявлялась выраженная патологическая неврологическая симптоматика (глазодвигательная, судорожный синдром т. д).
Также не выявлено патологических патернов при проведении амплитудинтегрированной электроэнцефалографии.
УЗИ органов брюшной полости: признаки расширения лоханок обеих почек.
Нейросонография: УЗИ-признаки незрелости структур головного мозга, асимметрия передних рогов боковых желудочков.
В расширенном метаболическом скрининге выявлен повышенный уровень иммунореактивного трипсина до 108 нг/мл (N 60,0).
Антропометрия. Окружность головы: при рождении 26 см, в 1 месяц 26 см, в 2 месяца 28 см.
Окружность груди: при рождении 24 см, в 1 мес. – 26 см, в 2 мес. – 28 см.
Масса тела: при рождении – 1130 г, в 1 мес. – 1520 г, в 2 мес. – 2040 г.
Длина: при рождении – 39 см, в 1 мес. – 42 см, в 2 мес. – 45 см.
Резюме: у ребенка отмечалось отсутствие роста окружности головы в первый месяц жизни, длительные апноэ на 3 месяце жизни, не позволяющие перевести ребенка на самостоятельное дыхание, отсутствие координации сосательного и глотательного рефлексов.
Патологоанатомический диагноз
- Основное заболевание:
а.Хромосомная патология (интерстициальная микроделеция 16 хромосомы); открытый артериальный проток (состояние после операции); микрокистоз почек, микроцефалия и микрогирия, стигмы дизэмбриогенеза.
b. Формирующаяся бронхолегочная дисплазия.
2. Осложнение: Облитерация полости перикарда, спайки в левой плевральной полости. Очаговые кровоизлияния в легкие.
3. Сопутствующие: Недоношенность.
Обсуждение
Известно, что не все внутриутробные состояния, включая генетически обусловленные, имеют выраженную эхографическую и биохимическую картину. Кроме того, «мягкие» УЗ-маркеры могут встречаться у плодов как с нормальным, так и с аномальным кариотипом.
Согласно приказу МОЗ Украины №641/84 от 31.12.2003 г., пациентка попадает в группу риска по возрасту и подлежит дальнейшему обследованию согласно результатам скрининга первого триместра. А именно: значительно отклонены ТВП плода (4,8 мм), b-ХГЧ (0,312 МОМ), РАРР-А (0,201 МОМ), пересмотренный риск трисомии 13/18: 1/2.
Имея рекомендацию к проведению инвазивной генетической диагностики, пациентка предпочла неинвазивный тест на наличие хромосомных аномалий плода (НИПТ) (PrenaTest), по которому получила отрицательный результат. Стоит отметить, что проведенный НИПТ позволял оценить возможность наличия хромосомных аномалий по 13, 18, 21, а также по половым хромосомам.
Отрицательное прогностическое значение (NPV), согласно расчету для каждой из исследованных хромосом, составило >99%. Однако, тест имеет ряд ограничений (KotsopoulouI, Tsoplou P, 2015:141-158) и не рассчитан на детекцию аномалий иных хромосом, включая микроструктурные аномалии.
Согласно рекомендациям Американского колледжа акушерства и гинекологии (ACOG) и Общества материнско-фетальной медицины, НИПТ-исследование может быть выбрано в качестве основной скрининговой стратегии (Committee opinion, 2015:1-6) безотносительно к величине индивидуального риска, и лишь при наличии порока развития целесообразно отдать предпочтение диагностическим инвазивным методам.
С другой же стороны, пациентам, которые проходят диагностику инвазивным путем при структурно нормальном плоде (по данным УЗИ), рекомендации не предусматривают предпочтение выбора ни в пользу кариотипирования, ни ДНК-диагностики микроструктурных хромосомных аномалий (методом сравнительной геномной гибридизации) (Committee opinion, 2013:1-4).
Одновременное существование таких рекомендаций кажется не вполне логичным в силу отсутствия связи с величиной индивидуального риска, поскольку последний рассчитывается на основании возрастного риска, модифицируемого индивидуальными результатами лабораторных исследований и УЗД. Таким образом, при определении стратегии исследования пациентки наличие порока развития плода, вынесенное в международных рекомендациях в виде основного критерия отбора, должно рассматриваться в комплексе с остальными. В противном случае алгоритм диагностики для пациентки с индивидуальным риском 1/2 (в нашем случае) фактически приравнен к таковому при 1/250.
Следуя рекомендациям доктора, пациентка проходит инвазивную диагностику (амниоцентез) с подтверждением нормального кариотипа и последующим УЗИ, выявившим признаки задержки развития плода. Со значительным опозданием обращается на проведение медико-генетического консультирования, которое позволило выявить дополнительные факторы риска. А именно: у супруги – смерть в раннем детстве родного брата в связи с врожденной аномалией кишечника и почек; у супруга в первом браке умер ребенок с пороком сердца.
Пара была осведомлена о возможности наличия микроструктурных хромосомных аномалий (5–6%) и приняла решение провести исследование криоконсервированныхамниоцитов методом чип-сравнительной геномной гибридизации (aCGH) для определения оптимальной стратегии сопровождения новорожденного, понимая, что вопрос прерывания беременности в случае идентификации хромосомной аномалии уже не может быть решен (после 22 недель). Результатом диагностики стала идентификация микроделеции 2,7 млн. п.о. в длинном плече хромосомы 16 (локус 16q22.1). 38% делеции перекрывало кодирующие регионы структурно значимых генов (рис. 1).
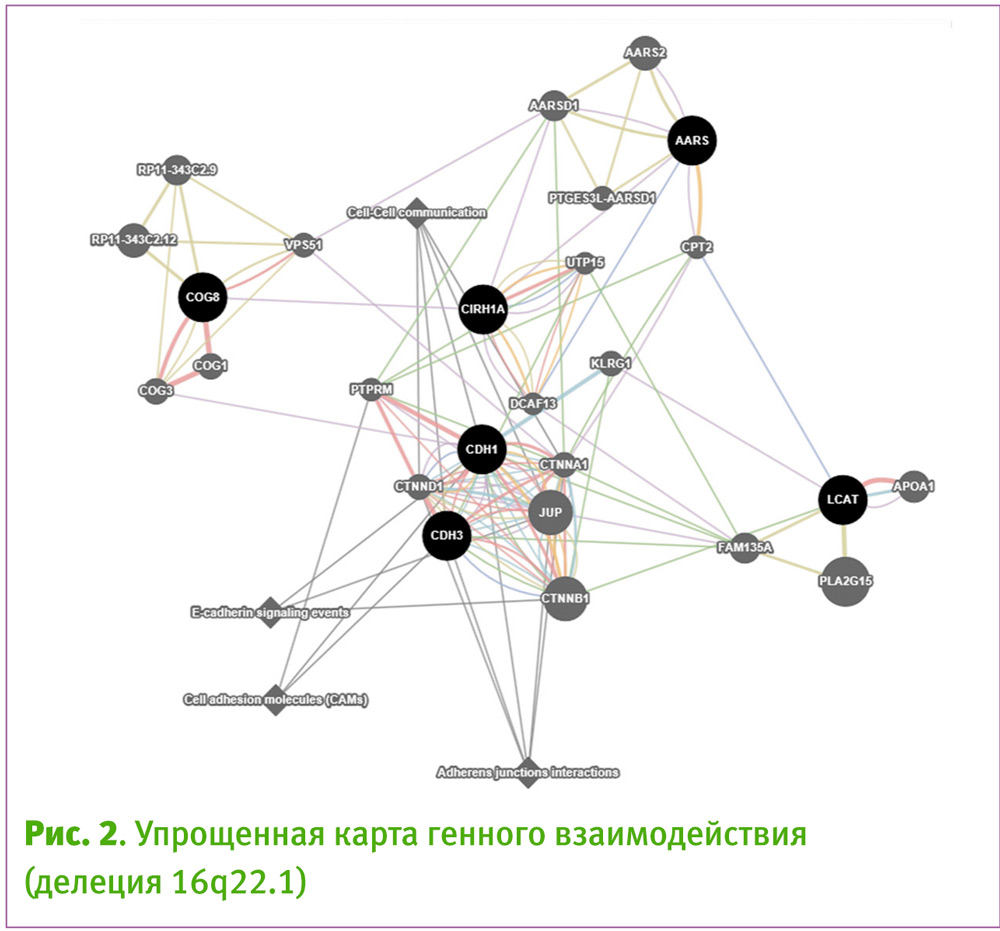
Виртуальная модуляция карты взаимодействия генов (рис. 2) свидетельствует о нарушенных физиологических каскадах, вовлекающих процессы межклеточного сигнального и контактного взаимодействия, а также обмена липидов.
Международная база данных Decipher на момент публикации содержала клиническую информацию о 32 пациентах со сходной микроделецией, из которых 10 были бессимптомными носителями, 22 имели клинические проявления различной степени. Патология проявлялась задержкой психомоторного развития, аутизмом, поведенческими/психиатрическими расстройствами в старшем возрасте. У большинства пациентов были врожденные аномалии (сердца, легочной артерии, дистальных отделов конечностей, костной системы, глазной щели, расщепление губы и неба)
Диагностика микроструктурных хромосомных аномалий одновременно в разрезе всего генома на сегодня возможна только методом сравнительной геномной гибридизации (Микитенко Д. О., Зукин В. Д., 2010: 183–187), так как новейшее решение в виде секвенирования «нового» поколения еще не отточено на определение протяженных делеций и дупликаций, захватывающих отдельные экзоны, гены и целые локусы.
Показанием для проведения исследования методом сравнительной геномной гибридизации может являться увеличение шейного пространства более 3,5 мм и/или порок развития, которые не сопровождаются хромосомными аномалиями при рутинном кариотипировании.
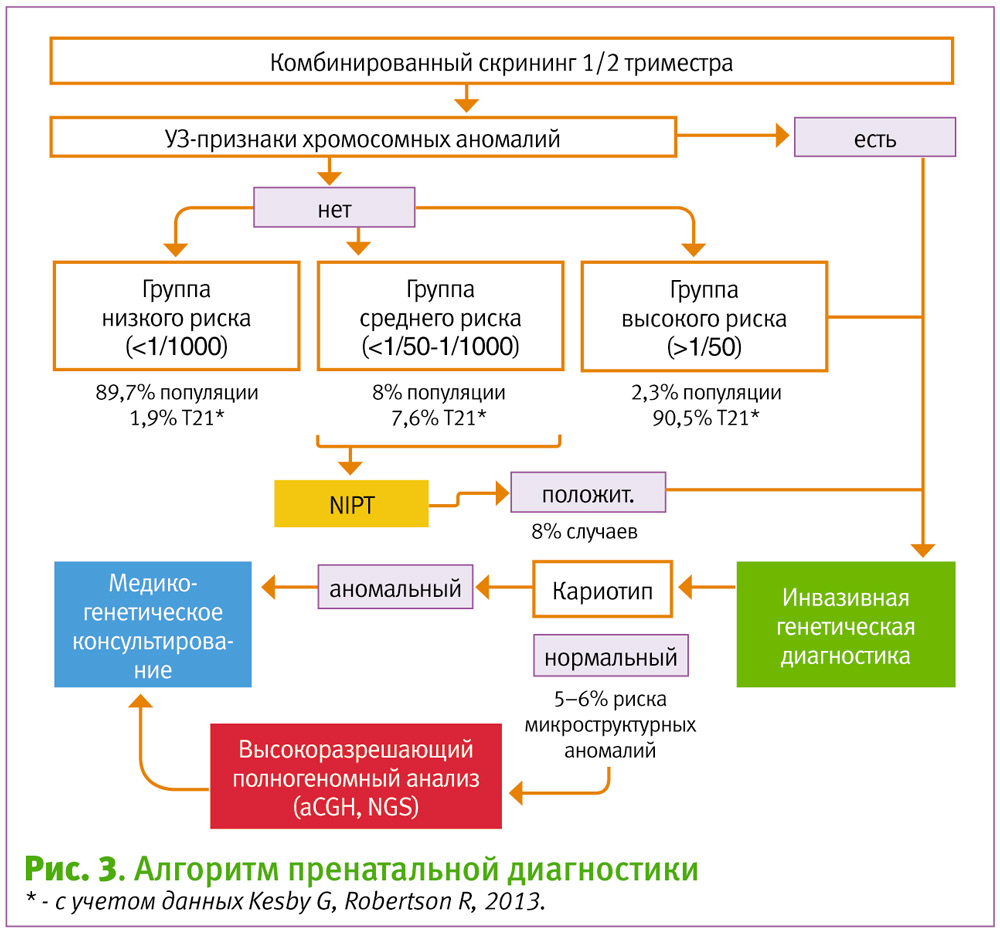
На этом основании целесообразно предложить следующий алгоритм пренатальной диагностики (рис 3).
Из предложенного алгоритма следует, что, несмотря на свою неточность, традиционные скрининговые исследования (с точки зрения cost effective) являются оптимальным отправным этапом пренатального исследования, так как позволяют определить группу риска. В случае высокого риска (более, чем 1/50) и/или визуализации УЗ-аномалии, целесообразно рекомендовать криотипирование плода путем инвазивной диагностики. Группа низкого риска (менее, чем 1/1000) не имеет медицинских показаний для дообследования. Риск наличия хромосомной аномалии плода в такой группе критически низкий. Группа беременных с риском 1/50–1/1000 требует особого подхода. Это обусловлено тем, что риск наличия хромосомной аномалии плода повышен, но он ниже шанса получить осложнение от инвазивной процедуры в виде прерывания беременности. Границы рисков группы (1/50–1/1000) обусловлены тем, что в среднем частота вынашивания беременности с хромосомной аномалией в этой группе около 7,6% и между крайними рисками отличается незначительно (Kesby G, Robertson R, 2013). Для этой группы наиболее оптимальной стратегией является проведение НИПТ. При положительном результате, целесообразно проводить инвазивную генетическую диагностику.
Однако, если при проведении определяется нормальный кариотип, и это сопровождается увеличением толщины ШП более 3,5 мм или эхо-признаками хромосомных аномалий, пациент должен быть предупрежден, что имеется риск микроструктурных хромосомных аномалий от 6% (Florentino F. et. al., 2013: 725–730) до 16% (LundI. C. et. al., 2015: 95–100). Это является основанием для продолжения диагностики с использованием высокоразрешающих методов полногеномного анализа – aCGH и/или NGS (секвенирование «нового» поколения) с последующим медико-генетическим консультированием.
Применение такого алгоритма обеспечит проведение точной генетической диагностики, которая является крайне важной для проведения медико-генетического консультирования, так как позволяет оценить прогноз состояния ребенка, решить вопросы планирования семьи. В семьях с высоким повторным риском целесообразно рекомендовать проведение пренатальной диагностики или преимплантационной диагностики эмбрионов в рамках цикла ВРТ для трансфера эмбриона в полость матки, заведомо не имеющего хромосомной аномалии.
Выводы
Применение арсенала современных молекулярно-диагностических подходов (НИПТ, aCGH) в пренатальной медицине позволяет пересмотреть алгоритм проведения дородовой генетической диагностики и существенно повысить эффективность выявления генетической аномалии.
С точки зрения эффективности использования ресурсов, применение этих методов является обоснованным в качестве «второй» линии диагностики в рамках предложенного алгоритма.
коментариев