

Введение
 По данным ВОЗ, в мире насчитывается около 250 млн. человек с нарушениями слуха, что составляет 4,2% от всей популяции земного шара.
По данным ВОЗ, в мире насчитывается около 250 млн. человек с нарушениями слуха, что составляет 4,2% от всей популяции земного шара.
Приблизительно 1 из 1000 детей имеет тяжелую или полную потерю слуха при рождении или в раннем детстве, что определяется как прелингвальная глухота.
Считается, что примерно 50–60% всех этих случаев обусловлено наследственными причинами. Оставшаяся часть приходится на неблагоприятные внешнесредовые влияния, оказываемые на органы слуха эмбриона или уже родившегося человека.
Генетические формы нарушения слуха встречаются чаще, чем другие виды врожденной наследственной патологии, такие как фенилкетонурия, гипотиреоз и муковисцидоз.
Традиционно врожденная тугоухость/глухота не считается тяжелым заболеванием, поскольку дети в большинстве своем соматически здоровы. Тем не менее, тяжелая потеря слуха обуславливает грубое нарушение речевого, психоэмоционального и социального развития, и является основанием для получения инвалидности.
Результативность терапии зависит от раннего выявления изменений слуха. Во всех случаях неэффективности консервативного лечения проводится слухопротезирование с помощью слуховых аппаратов, а в случаях глухоты – кохлеарная имплантация.
Прорыв в области молекулярно-генетических знаний об органе слуха выводит на качественно новый уровень подходы к установлению причин тугоухости и разработку эффективных способов лечения, а также открывает новые пути профилактики наследственных нарушений слуха.
Формы врожденных дефектов слуха
Врожденная и ранняя тугоухость проявляется двумя формами в зависимости от того, какой отдел органа слуха поврежден.
Различают три основных разновидности тугоухости: кондуктивную, нейросенсорную и смешанную.
Кондуктивная обусловлена поражением звукопроводящего аппарата внешнего и среднего уха. Такая форма поддается медикаментозному, или хирургическому лечению, или же легко исправляема с помощью несложного слухового аппарата, усиливающего звуки.
Причиной сенсоневральной тугоухости, или как ее еще называют, нейросенсорной, является нарушение преобразования механических колебаний в электрические импульсы, вызванная в большинстве случаев поражением волосковых клеток улитки уха, а также проводящих путей и коры головного мозга. Такая тугоухость необратима.
Смешанная форма сочетает в себе признаки двух вышеперечисленных, и представляет собой кондуктивную тугоухость с поражением внутреннего уха.
Тугоухость измеряется в децибеллах (dB). Слух считается нормальным, если слуховой порог данного индивидуума находится в пределах 0–15 dB от нормального порога слышимости. Степень тугоухости градуируется как: легкая (26–40 dB), умеренная (41–55 dB); умеренно тяжелая (56–70 dB), тяжелая (71–90 dB), глубокая (90 dB).
Наиболее частые негенетические причины врожденной и ранней глухоты
Основными негенетическими причинами развития дефектов слуха являются врожденные инфекции, токсическое воздействие лекарственных препаратов, гипоксически-асфиксические состояния во время беременности и родов.
Приобретенная потеря слуха у детей наиболее часто возникает в результате пренатальных TORCH-инфекций (токсоплазмоз, краснуха, цитомегаловирус, герпес) или постнатальных инфекций, в частности, бактериальных менингитов, вызываемых Neisseria meningitidis, Haermophilus influenzae или Streptococcus pneumoniae. Менингиты, вызванные многими другими организмами, включая Escherichia coli, Listeria monocytogenes, Streptococcus agalactiae и Enterobacter cloacae, также могут повлечь за собой потерю слуха. Асимптомная врожденная цитомегаловирусная инфекция часто не распознается и может быть ассоциирована с вариабельной флюктуирующей сенсоневральной потерей слуха.
Наследственные причины врожденной потери слуха
В клинической практике большое значение имеет медико-генетическое консультирование (при врожденных пороках слуха в том числе). Это связано с тем, что наследственные заболевания этиологически связаны с различными типами мутаций (генные, геномные, хромосомные).
Врачу-генетику необходимо применить синдромологический подход, определить фенотип, с участием сурдолога оценить степень поражения слуха, а также установить тип наследования, реккурентный риск, дать индивидуальный прогноз с применением всех доступных методов обследования и диагностики. Поскольку специалист, который не использует в своей практике доказательные данные, теряет возможность действенной помощи пациенту и рискует навредить здоровью.
Синдромальные повреждения слуха
Описано более 400 генетических синдромов, включающих потерю слуха. Синдромальные повреждения слуха составляют до 30% предречевой глухоты, но их относительный вклад по отношению ко всем случаям глухоты относительно невелик, что отражает проявление и диагностику постречевой потери слуха.
Несиндромальные повреждения слуха
По механизму наследования различают следующие типы:
- аутосомно-рецессивные (78%);
- аутосомно-доминантные (20%);
- х-сцепленные (1%);
- митохондриальные (1%).
Более 70% случав наследственной потери слуха – несиндромальные. Различные генные локусы обозначаются как DFN (от deafness – глухота). Локусы генов, наследуемые как аутосомно-доминантные, обозначаются как DFNA; такие же гены, наследуемые аутосомно-рецессивно, обозначаются как DFNB; гены, наследуемые сцепленно с Х-хромосомой – как DFN. В настоящее время известно более 100 генов, мутации в которых ответственны за нарушения слуха.
Семейные исследования аутосомно-доминантной несиндромальной потери слуха предполагают, что мутации в одном гене не отвечают за большинство случаев данного заболевания. Однако, отмечено, что аудиопрофиль может быть различным и прогнозируемым. Например, мутации в гене VFS1 обнаружены у 75% семей, в которых наследуется аутосомно-доминантное несиндромальное повреждение слуха, которое первично повреждает область низких частот, в то время как «спаривание» мутаций приводит к повреждению у потомства и восприятия высоких частот.
Аутосомно-рецессивные формы обычно более тяжелые, чем другие и почти все ведут к дефектам улитки (нейросенсорная глухота). Список генов, мутации в которых ответственны за развитие рецессивной глухоты, пополняется очень быстро, но, по данным многочисленных исследований, мутации гена GJB2 ответственны за более чем 50% случаев всей несиндромальной рецессивной патологии слуха во многих популяциях.
Х-сцепленное несиндромальное повреждение слуха.
Ген DFN3, картированный на Xq21.1, характеризуется кондуктивно-сенсоневральной потерей слуха, кондуктивный компонент которой вызывается неподвижностью стремечка. В противоположность другим типам кондуктивной потери слуха хирургическая коррекция противопоказана из-за аномальной коммуникации между цереброспинальной жидкостью и перилимфой, что приводит к просачиванию («перилимфатический фонтан») и полной потере слуха в случаях фенестрации овального окна или его удаления. Причиной болезни является ген POU3F4. Молекулярно-генетическое тестирование возможно на клиническом уровне.
Другие Х-сцепленные несиндромальные формы потери слуха включают глубокую прелингвальную потерю слуха, связанную с DFN2 и DFN4, начинающуюся также как DFN6, от 5 до 7 лет, двухстороннюю, высокочастотную, прогрессирующую вплоть до взрослого состояния, тяжелую вплоть до глубокой, на всех частотах. Глухота, связанная с DFN5, DFN7, DFN8 локусами, не описана.
Митохондриальное несиндромальное повреждение слуха.
Некоторые мутации митохондриальной ДНК вызывают несиндромальную потерю слуха. Описана гомоплазмическая мутация в nt1555 (A-G транзиция) в митохондриальном гене MTRNR1 в двух семьях. Эта же мутация была обнаружена у людей с аминогликозид-индуцированным ототоксическим повреждением слуха. В двух других семьях с наследуемой по материнской линией несиндромальной потерей слуха были идентифицирована гетероплазмия по A-G транзиции в nt7445 в гене MTTS1. Пенетрантность гена этой формы потери слуха, вызываемой митохондриальными мутациями, была очень низкой, что заставляет предполагать существование неидентифицированных генетических или средовых факторов, играющих роль в прогрессирующем повреждении слуха.
Большая часть случаев генетической глухоты исходит из мутаций, вовлекающих один ген. В то же время идентифицируются небольшое и растущее количество случаев, при которых потеря слуха происходит при мутациях в двух независимых генах. Среди браков глухих людей, имеющих в родословных глухоту в нескольких поколениях, собираются вместе редкие гены глухоты всех типов. В частности, причиной большого распространения глухоты от мутаций в GJB2 считают введение 400 лет тому назад в западных странах языка жестов. Наступившая при этом лингвистическая гомогамия, когда выбор брачных пар основан на способе общения, способствовали возникновению таких браков и удвоению частоты глухоты по GJB2 (диаграмма 1).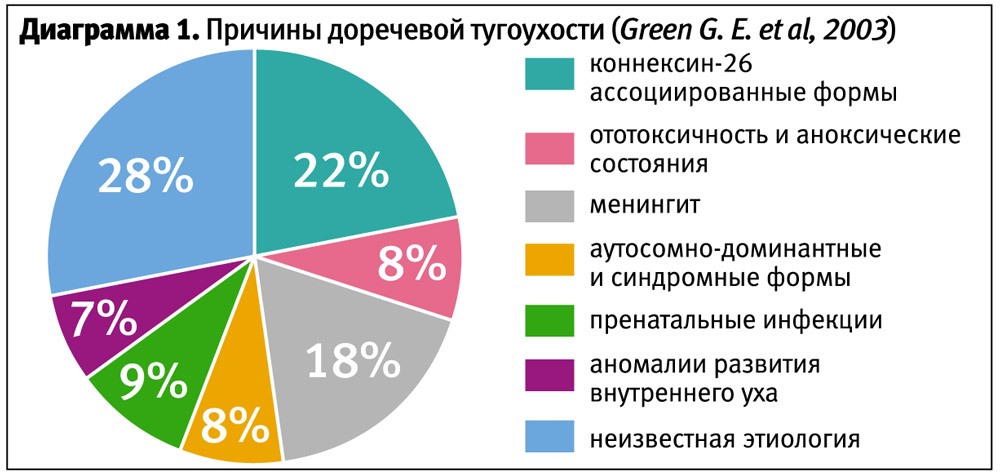
Мутации гена коннексина-26 (GJB2) – основная причина наследственной глухоты
Наиболее важный локус для несиндромальной аутосомно-рецессивной глухоты (DFNB1) первоначально был найден на хромосоме 13q11 путем анализа сцепления в двух крупных близкородственных тунисских семьях с доречевой, выраженной глухотой. Последующие исследования сцеплений в новозеландской/австралийской и итальянской/испанской семьях с глухотой показали, что этот локус является главным вкладчиком в доречевую глухоту. Мутации в гене GJB2, кодирующем белок щелевых соединений коннексин-26, который был картирован в 13q11-q12, затем были идентифицированы в трех родственных семьях в Пакистане с выраженной глухотой, генетически сцепленной с 13q11. Ген GJB2 – первый DFNB-ген – идентифицирован в 1997 году.
В настоящее время известно более 100 различных мутаций в гене GJB2, большинство из которых этнически специфичны. Наиболее распространенными из них являются рецессивные мутации: делеции 35delG, 167delT и 235delC, и замены R143W, W24X.
В то время как ген GJB2 является главным геном, ответственным за несиндромальную рецессивную глухоту во многих популяциях, имеются некоторые противоречия относительно роли GJB2 в доминантной глухоте. Некоторые мутации гена GJB2 в гетерозиготном состоянии, кодирующие определенный домен белка (первый внеклеточный домен), как установлено, сегрегируют с аутосомно-доминантной потерей слуха в небольшом количестве семей с разными фенотипами, с началом в позднем детском возрасте, от слабой до выраженной прогрессирующей.

Установлен эффект основателя для мутации 35delG у населения Ближнего Востока, Европы и Северной Америки (возраст мутации оценивается » в 10 000 лет). Эффект основателя также показан для мутаций 235delC в популяциях Восточной и Центральной Азии (Япония, Корея, Китай, Монголия, возраст мутации » 11 500 лет) и W24X – в Индии (возраст мутации » 7880 лет). Мутация 167delT зарегистрирована с высокой частотой среди евреев ашкенази.
Одна из специфических мутаций 35delG гена GJB2, выявляемых в популяциях европеоидной расы, представляет собой одну из наиболее частых мутаций, вызывающих болезнь. Частота носительства 35delG мутации составляет 3,5–4,0% в итальянской и греческой популяциях, это приводит к тому, что гомозиготность по этой мутации встречается с частотой 1 на 2500 новорожденных в этих этнических группах.
Фенотипические проявления мутации 35delG гена GJB2
Мутация 35delG представляет собой делецию гуанина (G) в положении 30–35-й последовательности гена, что приводит к сдвигу рамки считывания, образованию преждевременного стоп-кодона и синтезу аномального белка коннексина-26.
Последний представляет собой трансмембранный транспортный белок, шесть субъединиц которого формируют цилиндрические межклеточные щелевые контакты между соседними клетками, образуя каналы для пассивного транспорта молекул и электролитов.
Мутации гена GJB2 вызывают патологические изменения, приводящие к синтезу дефектного белка коннексина-26 и, в дальнейшем, к затруднению транспорта электролитов, АТФ и глюкозы между клетками. В результате этих изменений происходит нарушение рециркуляции ионов К+, которое приводит к гибели сначала поддерживающего, а затем волоскового нейроэпителия спирального органа.
Гомозиготное носительство этих мутаций проявляется глубокой двусторонней тугоухостью или полной глухотой, развивающейся в периоде до 1 года. Ребенок с такой патологией не имеет внешних дефектов слуховой системы, т. е. мутация клинически проявляется только симметричным и необратимым снижением слуха. Гетерозиготное носительство 35delG не приводит к снижению слуха.
Коннексин-26 обнаружен во всех органах и тканях, но его изменения в значительной степени отражаются только на органе слуха. Отсутствие поражения других органов объясняют тем, что во внутреннем ухе он не может быть замещен на другой вид коннексина, как это происходит во многих тканях.
Скрининг патологии слуха у новорожденных
Распространенность дефектов слуха у детей и поздняя их диагностика всегда препятствовали проведению ранних реабилитационных мероприятий, что требовало внедрения скринирующих программ среди новорожденных.
Внедрение скрининга произошло в 1989 году в США. А в 1993 году Национальный институт здоровья США пришел к выводу, что аудиологический скрининг должен быть применен для всех новорожденных в течение первых трех месяцев жизни ребенка. И в 2004 г. в странах, внедривших такой скрининг, регистрировался охват от 93 до 99% детей. Средний возраст, при котором диагностируется потеря слуха, снизился с 24–30 месяцев до 2–3 месяцев.
В настоящее время разработаны и используются в практической деятельности ряд объективных диагностических методов определения слуха у новорожденных: акустическая импедансометрия, аудиометрия с регистрацией различных классов слуховых вызванных потенциалов, вызванная отоакустическая эмиссия, задержанная отоакустическая эмиссия (ЗВОАЭ).
Последний метод получил предпочтение в качестве метода скрининга новорожденных. Эффективность такой методики составляет 85% (имеет диагностическое знание лишь при тяжелых степенях тугоухости/глухоты).
ЗВОАЭ представляет собой ответный звуковой сигнал, возникающий на 8–12 мс после включения стимуляции и продолжающийся 10–30 мс. Для регистрации используют вводимый в наружный слуховой проход зонд, в корпусе которого размещены миниатюрный телефон и микрофон. Стимулами служат широкополосные акустические щелчки с частотой повторения 20–50 с.
Второй этап скрининга – методика коротколатентных слуховых вызванных потенциалов (КСВП).
Предполагается, что часть гомозиготных новорожденных носителей 35delG имеет клинически сохранный слух, проходя при этом аудиотест и имея незначительные изменения показателей амплитуды отоакустической эмиссии. В ряде европейских стран (Греция, Испания, Италия) и некоторых штатах США это послужило причиной для проведения обязательного молекулярно-генетического скрининга «мажорных» мутаций в гене коннексина-26 параллельно с аудиологическим скринингом новорожденных.
Проведение совместного аудиологического и молекулярно-генетического скрининга в этих странах показало, что часть из таких детей может быть не выявлена при проведении аудиологического скрининга, поскольку нарушение слуха у них на момент рождения не выражено или выражено незначительно.
Возможности проведения последовательного аудиологического и молекулярно-генетического скрининга патологии слуха в Украине
Учитывая вероятность высокой распространенности мутаций гена коннексина-26 и среди населения Украины, в Межобластном центре медицинской генетики и пренатальной диагностики (Кривой Рог) во втором полугодии 2011 года была предложена, апробирована (при сотрудничестве с сурдологами Днепропетровской, Черкасской, Кировоградской и Запорожской областей), введена в практику и запатентована модель обеспечения молекулярно-генетического обследования новорожденных после аудиологического скрининга.
По результатам аудиологического скрининга сурдологами в генетический центр подаются данные о детях, у которых обнаружены врожденные нарушения слуха. Эти дети обследуются на предмет моногенной этиологии тугоухости и глухоты (мутация 35delG гена GJB2). Материалом для исследований являются сухие пятна крови на тест-бланках, которые были присланы для неонатального скрининга на распространенные наследственные заболевания (фенилкетонурия, врожденный гипотиреоз, муковисцидоз и адреногенитальный синдром), проводимый в этом же учреждении. Все данные после исследований направляются к сурдологу для формирования группы с подтвержденной рецессивной нейросенсорной глухотой и подбора индивидуальной тактики коррекции слуха.
По результатам молекулярно-генетического исследования семьи, у которых выявлена моногенная природа заболевания, вызываются на консультацию к врачу-генетику для постановки на учет, обследования всех членов семьи и проведения ДНК-диагностики при последующих беременностях.
При применении вспомогательных репродуктивных технологий также возможно проведение пренатального исследования (в числе других) по наличию мутантных аллелей в гене коннексина-26 на стадии преэмбриона, подготовленного для трансфера.
Случаи из практики
- Областным сурдологом Днепропетровской области в Межобластной центр медицинской генетики и пренатальной диагностики (Кривой Рог) была подана информация о группе детей с врожденными дефектами слуха. Среди других случаев моногенная природа болезни была подтверждена у девочки А. Ребенок с родителями был направлен на консультацию к врачу-генетику и для обследования семьи. Помимо родителей ребенка, были обследованы также сестра отца ребенка и члены ее семьи на предмет носительства мутантного гена. В семье сестры носительство мутации не обнаружено. Родителям больного ребенка была рекомендована пренатальная диагностика на предмет глухоты в случае последующих беременностей. При наступлении беременности родители отказались от дородовой диагностики. Ими было принято решение провести обследование после рождения параллельно с неонатальным скринингом на наследственные заболевания. По результатам ДНК-диагностики родившийся ребенок оказался гетерозиготным носителем мутантного аллеля.
- К врачу-генетику обратились родители ребенка С. (2 года), которому сурдологом был выставлен диагноз «нейросенсорная тугоухость III степени». При этом ребенок в роддоме прошел аудиологический скрининг и не было выявлено нарушения слуха. Основной причиной потери слуха считалась перенесенная ГРВИ. Этой семье было проведено исследование по выявлению мутации гена коннексина-26. Моногенная природа болезни подтверждена. Через полгода семья обратилась за консультацией по поводу проведения пренатальной диагностики. Для забора плодного материала применен амниоцентез в сроке 20 недель. Проведенное исследование показало, что плод также имеет гомозиготный генотип по мутации. Беременность закончилась родами в 40 недель рождением мальчика. После рождения семьей безотлагательно приняты меры по коррекции слуха.
- К врачу-генетику обратилась семья Д. с двойней (возраст 2,5 года), причем мальчику поставлен диагноз нейросенсорная глухота, а девочка была здорова. После проведенной ДНК-диагностики у мальчика наследственная природа заболевания подтверждена, а девочка оказалась только носителем, что является редким явлением. При последующих беременностях семье рекомендована пренатальная диагностика.
Заключение
Учитывая современные методы реабилитации слабослышащих и глухих детей (слухопротезирование, кохлеарная имплантация), сегодня проблемой сурдологии является не столько сама патология слуха, сколько ранняя диагностика слуховых нарушений, так как позднее обнаружение становится причиной невосполнимого постнатального недоразвития слуховой коры. В этих случаях даже самые современные системы электроакустической стимуляции неспособны оказать свой реабилитационный эффект. Поэтому столь важной оказывается ранняя диагностика и своевременная реабилитация детей с доречевыми формами патологии слуховой системы. Также ставится вопрос о возможности в раннем периоде новорожденности (учитывая молекулярно-генетические механизмы патологии слуха) приостановить или затормозить прогрессирующее снижение слуха и не допустить, или отсрочить развитие глубокой степени тугоухости у пациентов с генетическими мутациями.
Генная терапия могла бы решить все эти проблемы, однако она пока является перспективой далекого будущего. Решением ближайшего времени может стать разработка концепции геноспецифической слухосохраняющей фармакотерапии, исходя из современных представлений о молекулярно-генетическом патогенезе различных форм генетических нарушений слуха.
Предлагаем сотрудничество с другими регионами с возможностью дистанционного обследования пациентов, имеющих врожденные нарушения слуха.
ОКУ «Межобластной центр медицинской генетики и пренатальной диагностики»
50000, г. Кривой Рог,
пл. Освобождения, 3-А, тел./факс:
(0564) 92-49-30; 92-42-73;
e-mail: genetika@ukrpost.ua
лаборатория молекулярной генетики: тел. 0564 92-36-19;
e-mail: genetika7@gmail.com
коментарів