

Синдром Марфана (СМ) редко диагностируется у новорождённых [2]. Младенческий СМ отличается по клиническим и морфологическим признакам от классических его проявлений у юношей и взрослых. При данном варианте течения СМ основную опасность представляет поражение сердечно-сосудистой системы, в 50% случаев заканчивающееся гибелью пациентов [1, 2, 4].
Целью настоящего анализа является демонстрация клинического случая генетически обусловленной патологии соединительной ткани — синдрома Марфана у новорождённого ребёнка.
Мальчик К., рождённый от ІІІ беременности (первая завершилась самопроизвольным абортом в 6 недель гестации, вторая — рождением, со слов мамы, здорового ребёнка), протекавшей с гестационными отёками, на фоне хронического гастрита, дисплазии шейки матки. У отца ребёнка марфаноподобный фенотип — высокий, худощавый, с узким лицом, длинными конечностями, арахнодактилией, воронкообразной деформацией грудной клетки, кифозом и врождённым пороком сердца (тетрада Фалло).
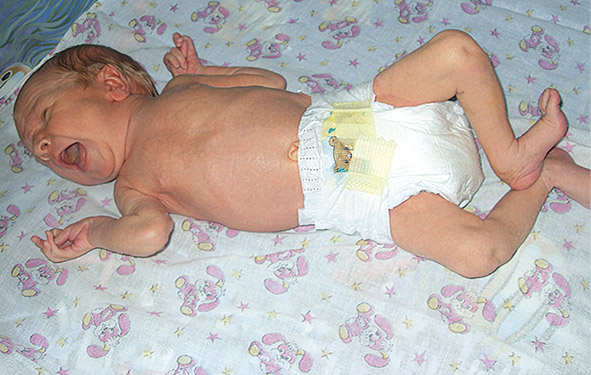 Роды ІІ, в 39–40 недель, околоплодные воды светлые, умеренное количество, течение периодов родов без особенностей. Масса тела ребёнка при рождении 3250 г, длина тела — 55 см, окружность головы — 33 см, окружность грудной клетки — 33 см; оценка по шкале Апгар 7–8 баллов. Ребёнок имел выраженные стигмы дизэмбриогенеза, мышечную гипотонию, снижение активности рефлексов, синдром желтухи с 4-х суток жизни, негрубый систолический шум над сердцем. На 6-е сутки жизни мальчик переведен в отделение патологии новорождённых. Стигмы дизэмбриогенеза включали (фото 1): астеническое телосложение, длинные конечности, арахнодактилию, положительный тест запястья (при обхвате большим пальцем и мизинцем ребенка запястья его другой руки — их концевые фаланги накладываются); положительный тест большого пальца (фиксация большого пальца поперёк ладони без дополнительной помощи — ногтевая фаланга большого пальца выходит за ульнарный край ладони) — фото 2; невозможность полного разгибания рук в локтевых суставах — разгибание менее 170° — фото 3.
Роды ІІ, в 39–40 недель, околоплодные воды светлые, умеренное количество, течение периодов родов без особенностей. Масса тела ребёнка при рождении 3250 г, длина тела — 55 см, окружность головы — 33 см, окружность грудной клетки — 33 см; оценка по шкале Апгар 7–8 баллов. Ребёнок имел выраженные стигмы дизэмбриогенеза, мышечную гипотонию, снижение активности рефлексов, синдром желтухи с 4-х суток жизни, негрубый систолический шум над сердцем. На 6-е сутки жизни мальчик переведен в отделение патологии новорождённых. Стигмы дизэмбриогенеза включали (фото 1): астеническое телосложение, длинные конечности, арахнодактилию, положительный тест запястья (при обхвате большим пальцем и мизинцем ребенка запястья его другой руки — их концевые фаланги накладываются); положительный тест большого пальца (фиксация большого пальца поперёк ладони без дополнительной помощи — ногтевая фаланга большого пальца выходит за ульнарный край ладони) — фото 2; невозможность полного разгибания рук в локтевых суставах — разгибание менее 170° — фото 3.
При клиническом осмотре тяжесть состояния определяли неврологические расстройства (ограничение общей и рефлекторной активности, общая скованность, отсутствие эмоций голода и дискомфорта), дыхательные расстройства (поверхностное тахипноэ до 80 в 1 минуту, тахикардия до 160 в 1 минуту), синдром желтухи. Со стороны других внутренних органов патологии не выявлено. Динамическое наблюдение офтальмологом позволило только в 2-месячном возрасте диагностировать подвывих хрусталика. При проведении дополнительных методов исследования было выявлено наличие искривления позвоночника и диафрагмальной грыжи справа (фото 4, 5).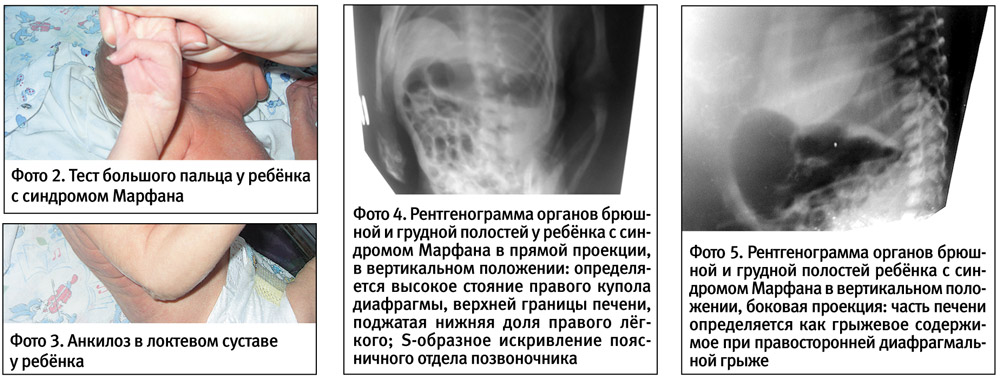
При ультразвуковых исследованиях выявлены следующие изменения:
- со стороны головного мозга — признаки незрелости (или мальформации): открытая полость прозрачной перегородки и полость Верге в сочетании с двусторонними субэпендимальными кровоизлияниями;
- со стороны сердца — дилатация корня аорты (дилатация в области синусов 17,7 мм, восходящей части — 9,3 мм), регургитация на трикуспидальном клапане ІІІ степени, на митральном клапане — ІІ степени, а также регургитация в створках клапанов лёгочной артерии и аорты, пролабирование межпредсердной перегородки, функционирование овального окна;
- со стороны печени — изменение формы органа за счёт выступающего кверху участка;
- со стороны остальных внутренних органов патологии не выявлено.
При проведении ЭКГ регистрировалась неполная атриовентрикулярная блокада, дисметаболические изменения фазы реполяризации желудочков с депрессией сегмента S-T.
Электроэнцефалография: низкоамплитудная неустойчивая полиритмичная активность. Топическая организация отсутствует. Фазовая организация сна проявляется слабо. Специфические эпифеномены не выявлены.
При проведении лабораторных методов исследования было выявлено:
- в общих анализах крови и мочи — норма;
- функция почек не нарушена;
- непрямая гипербилирубинемия без повышения АлАТ и тимоловой пробы;
- снижение уровня холестерина плазмы (2,1 ммоль/л);
- преходящая гипокортизолемия;
- функция щитовидной железы — в норме;
- снижение уровня Т-лимфоцитов, относительное повышение уровня Ts, сопровождавшееся снижением показателя Th/Ts, снижение титра общих At уровня IgG.
В динамике у ребёнка прогредиентно нарастали дыхательные расстройства (нарастание поверхностного тахипноэ), НК усилилась до ІІ-А ст., формировался синдром задержки статико-кинетического развития.
Наличие диафрагмальной грыжи обусловило необходимость проведения оперативного лечения в 2-месячном возрасте. В послеоперационном периоде у мальчика наросла сердечно-сосудистая недостаточность, на фоне которой развился отёк лёгких, застойная пневмония, отёк головного мозга с последующей полиорганной декомпенсацией, приведшей к гибели ребёнка.
В отделении проводилась дифференциальная диагностика с другими соединительно-тканными заболеваниями: синдромом Ашара, Стиклера, Шпрентзена-Гольбера, элерсоподобными состояниями, MASS, синдромом Билса. Сочетание имеющихся больших и малых клинических, ультразвуковых и рентгенологических признаков, а также семейного анамнеза, позволило диагностировать у ребёнка синдром Марфана с указанием выявленных пороков развития.
Патологоанатомическое исследование подтвердило, что основной причиной смерти явилась врожденная патология сердечно-сосудистой системы как составная часть синдрома Марфана. Выявлена шаровидная кардиомегалия 6×6×5 см, масса сердца 55,0 г (при норме 27,0 г); резкая дилатация полостей сердца; папиллярные мышцы левого и правого желудочков истончены; эндокард на всём протяжении белесовато-серый, миокард очень дряблый, тусклый, на разрезе цвета вареного мяса; толщина стенки левого желудочка 0,3 см, правого — 0,3 см (при норме 0,5 см). Определяется расширение просвета всех клапанов, створки не смыкаются, свободно пролабируют в полость обоих предсердий и просвет аорты; створки клапанов истончены, хорды также истончены, в виде сероватых тяжей. Просвет восходящей части аорты расширен. При гистологическом исследовании стенки сердца определяется разволокнение, местами фрагментация миокардиоцитов; в интерстиции и периваскулярно — разрастание рыхло-волокнистой соединительной ткани. В восходящей части аорты определяется эндотелий с дистрофическими изменениями, средняя оболочка с выраженными дистрофическими изменениями плотно-волокнистой соединительной ткани, вплоть до некробиоза и некроза с формированием мелких щелевидных кистозных полостей.
Синдром Марфана относится к фибриллинопатиям — поражению соединительной ткани, являющимся результатом мутации в генах FBN1, FBN2, FBN3, что определяет клиническое разнообразие форм — от стёртых до тяжёлых [1, 3, 4].
СМ — заболевание соединительной ткани с поражением многих органов и систем, с преимущественной локализацией в костно-суставной, сердечно-сосудистой системах, глазах. При этой патологии соединительная ткань имеет повышенную растяжимость и недостаточную устойчивость к физическим нагрузкам. [1, 2, 3]. Тип наследования — аутосомно-доминантный. Распространенность СМ составляет 2–3 случая на 10 тысяч населения [1, 4, 5]. 75% случаев болезни передаются генетически и 25% выявляются как спорадические мутации.
В основе дефекта соединительной ткани лежит нарушение синтеза белка — фибриллина, являющегося важным компонентом эластических волокон. При этом происходят изменения в срединном слое сосудистой оболочки, характеризующиеся разрушением эластического каркаса и фрагментацией эластических волокон, нарушением расположения коллагеновых волокон, дистрофией гладкомышечных клеток, накоплением между волокнистыми структурами мукополисахаридов и формированием кист [1, 4, 5].
Клинические проявления СМ как одного из вариантов системной дисплазии соединительной ткани охватывают внешние проявления, изменения внутренних органов и глаз.
Диагноз СМ устанавливается на основании критериев Ghent, принятых в 1996 году [3].
Заболевание диагностируется по наличию больших и малых клинических признаков: наличие одного большого критерия в двух системах и одного малого — в третьей.
Большие критерии включают следующие проявления:
а) поражение опорно-двигательного аппарата — арахнодактилию, контрактуры в локтевом суставе, отношение длины верхнего сегмента тела к нижнему <0,86 (у взрослых) или размах рук/рост <1,05, килеподобную или воронкообразную деформацию грудной клетки ІІІ-ІV ст.; сколиоз ІІІ-ІV ст.; плоскостопие, протрузию acetabuli;
б) сердечно-сосудистой системы — дилатацию корня аорты, расслоение аневризмы аорты, расширение синуса Вальсальвы;
в) со стороны глаз — вывих (подвывих) хрусталика;
г) эктазию твёрдой мозговой оболочки, спинномозговую грыжу, рахисхиз позвонков.
К малым критериям относятся:
а) со стороны опорно-двигательного аппарата — воронкообразная деформация грудной клетки І-ІІ ст., гипермобильность суставов, черепно-лицевые дизморфии (долихоцефалия, узкое «птичье лицо»), высокое нёбо;
б) со стороны сердечно-сосудистой системы — дилатация лёгочной артерии, пролапс митрального клапана; дилатация других сосудов;
в) со стороны глаз — миопия, уплощение роговицы, гипоплазия радужки;
г) со стороны бронхолёгочной системы — пневмоторакс, эмфизема лёгких;
д) со стороны кожи — стрии.
Подтверждением диагноза являются генетические исследования — выявление мутаций в гене FBN1 в аутосоме 15q 21.1, наличие фенотипических проявлений у родителей и родственников.
Внешний вид больного с СМ достаточно характерен — узкий лицевой череп, высокий рост, длинные конечности с паукообразными пальцами, астеническое телосложение, нарушение функций суставов. Сердечно-сосудистые аномалии при СМ чаще всего включают поражение стенок аорты, лёгочной артерии, незаращение артериального протока [1, 2, 3, 4, 5]. Изменения твёрдой мозговой оболочки и генетические признаки являются дополнительными критериями.
Неонатальный СМ представляет собой сочетание арахнодактилии, внешних дисгармонических черт развития, пролапса обоих атриовентрикулярных клапанов и дилатации аортального и лёгочного корней. Морфологически находят миксоматозные наложения на клапанах, аневризму Вальсальвы, миксоматозную ткань вокруг атриовентрикулярного узла [2].
За 35-летний опыт работы отделения патологии новорождённых ДГКБ №3 г. Днепропетровска (20208 новорождённых пациентов) СМ наряду с другими врождёнными синдромами регистрировался реже, чем в 1% случаев [6].
Заключение
Данный клинический пример демонстрирует раннее и полисистемное поражение соединительной ткани с неблагоприятным исходом. Наличие диафрагмальной грыжи утяжеляет течение заболевания, а оперативное вмешательство несёт большой риск развития осложнений со стороны сердечно-сосудистой системы.
Литература
- Зербіно Д. Д., Ольхова О. В., Жураєв Р. К. Синдром Марфана: історичний ракурс і сучасний погляд на етіологію, патогенез, діагностику, клініку та лікування // Укр. мед. часопис. — 6 (80).— XІ\XІ. 2010. — С.97.
- Неонатальная кардиология / А. В. Прахоа.— Н. Новгород: Издательство Нижегородской госмедакадемии, 2008.— С. 355.
- De Poepe A., Devereux R. B., Dietz H. C. el al. Revised diagnostic criteria for the Marfan syndrome. Am I Mtd Genet 1996, 62: 417–26.
- Наследственные синдромы и медико-генетическое консультирование: Справочник / Козлова С. И., Семанова Е. М., Демикова Н. С., Блинникова О.Е. — Л.: Медицина, 1987.—С. 110.
- Барашнев Ю. И., Бахарев В. А., Новиков П. В. Диагностика и лечение врождённых и наследственных заболеваний у детей. — М.: «Триада-Х», 2004. — С. 113.
- Досвід роботи відділення патології новонароджених дитячої клінічної міської лікарні №3 м. Дніпропетровська за 35 років / Бєлозьорова В. Л., Бондаренко М. П., Жоголєва С. Ю., Федотова І. Б., Дробич С. О., Токарєв Д. С., Бігма Я. О. // Медичні перспективи. — 2011, Т. XVІ. — №2, ч.2. — С.14.
коментариев