

Впервые термин «орфанные болезни» (orphan – сирота, англ.) по отношению к редким заболеваниям появился в 1983 г. в США, а уже в 2002 г. в этой стране был принят Акт о редких заболеваниях (Rare Disease Act), который определял редкие болезни как «болезни или состояния, затрагивающие менее 200 000 людей в США». В Евросоюзе к редким заболеваниям относят болезни с распространенностью не более 5 случаев на 10 тысяч человек, в Японии — 1 человек на 50 тысяч, в Австралии — 1 человек менее чем на 2000 жителей страны
В Украине в 2014 году принят Закон №1213-VII, который позиционирует редкое (орфанное) заболевание как заболевание, которое угрожает жизни человека, или которое хронически прогрессирует, приводит к сокращению продолжительности жизни гражданина или к его инвалидности, и распространенность которого среди населения не превышает 1:2000.
Ввиду своей редкой встречаемости среди населения, орфанные болезни зачастую не учитываются врачами при проведении дифференциальной диагностики. Также сложность заключается в том, что диагностика данной патологии не является прерогативой врачей одной специальности, а требует участия специалистов различных сфер: гематологов, гастроэнтерологов, ревматологов, психоневрологов, невропатологов и особенно педиатров.
В то время, как педиатры научились понимать, распознавать и лечить инфекционные и соматические заболевания у детей, генетические болезни и онкологические заболевания в детском возрасте остаются terra incognita. Изучение редких заболеваний на сегодняшний момент, пожалуй, – одно из главных направлений педиатрии.
В большинстве случаев редкие заболевания — это хронические болезни, требующие сложной и дорогостоящей диагностики, а также пожизненного лечения.
Поздняя диагностика и отсутствие лечения приводят к той или иной степени инвалидизации пациента, снижают качество и продолжительность его жизни.
Лизосомные болезни накопления (ЛБН) – это общее название для группы различных заболеваний, характеризующихся генетической гетерогенностью и выраженным разнообразием клинических проявлений. Молекулярные механизмы развития ЛБН сходны. Все они обусловлены генетическими изменениями лизосомальных ферментов, которые контролируют процесс внутриклеточного расщепления таких макромолекул как гликозаминогликаны, гликолипиды, гликопротеины. Лечение данных болезней до недавнего времени было направлено на устранение симптомов заболевания и являлось, по сути, паллиативным, однако сейчас появилась возможность использования заместительной терапии с использованием искусственно разработанного недостающего фермента (т. н. рекомбинантного фермента).
Введение недостающего фермента предупреждает накопление патологического субстрата в клетках организма и позволяет избежать необратимых изменений в различных клетках и органах.
Лечение является пожизненным и должно быть начато как можно раньше. Однако раннее начало лечения возможно только при ранней диагностике заболевания.
К сожалению, многообразие симптомов, малое распространение ЛБН и недостаточная осведомленность медицинской общественности о данной проблеме приводят к поздней диагностике и, соответственно, к позднему назначению заместительной терапии и, в лучшем случае, к незначительному увеличению продолжительности жизни пациента и улучшению качества его жизни.
Болезнь Фабри относят к редким генетическим заболеваниям, поскольку ее распространенность, по данным международных исследований, колеблется от 1:40 000 до 1:117 000 среди мужчин и 1:20 000 среди женщин. Однако, по результатам массового скрининга новорожденных, проведенного в Италии, частота заболевания составила 1:3100 детей, что говорит о наличии большого количества недиагностированных форм болезни Фабри.
Болезнь Фабри относится к лизосомным болезням накопления и связана с нарушениями метаболизма сфинголипидов. Заболевание имеет тяжелое, прогрессирующее течение и фатальный исход. В большинстве случаев первые клинические проявления заболевания возникают в возрасте 20–30 лет, а средняя продолжительность жизни пациента составляет 50–60 лет даже при проведении паллиативного лечения.
Впервые болезнь Фабри была описана в 1898 г. двумя дерматологами – Джоном Фабри (J. Fabry) из Германии и Вильямом Андерсоном (W. Anderson) из Англии независимо друг от друга, поэтому второе, менее распространенное, название заболевания – болезнь Андерсона–Фабри.
Основой развития заболевания является мутация гена GLA, расположенного на длинном плече Х-хромосомы Хq22.1, вызывающая недостаточность a-глюкозидазы А и накопление субстрата – нейтральных гликосфинголипидов, главным образом – глоботриаозилцерамида (GL-3). Болезнь Фабри относится к заболеваниям, сцепленным с Х-хромосомой. Чаще болеют мужчины, однако женщины, которые являются носительницами мутантного гена, могут иметь клиническую картину заболевания не менее тяжелую, чем мужчины.
Дефекты гена GLA чрезвычайно разнообразны. Болезнь Фабри является результатом многочисленных миссенс- или нонсенс- точечных мутаций, которых в настоящее время описано более 600. Большинство мутаций уникальны для каждой отдельной родословной.
Недостаточность лизосомального фермента альфа-галактозидазы А вызывает аккумуляцию GL-3 в лизосомах разных тканей человеческого организма и физиологических жидкостях.
У здорового человека GL-3 образуется в результате распада стареющих эритроцитов и под действием глюкозидазы расщепляется на лактозу и лактазилцерамид. У пациента с болезнью Фабри снижение/отсутствие активности фермента приводит к накоплению GL-3 в эндотелии кровеносных сосудов, ганглиях вегетативной нервной системы, капиллярах и эпителиальных клетках почечных клубочков и канальцев, кардиомиоцитах, а также в роговице, гистиоцитарных и ретикулярных клетках соединительной ткани, что инициирует развитие воспаления и, в дальнейшем, фиброза, приводя к нарушению функции большинства органов и систем.
Первые признаки классической болезни Фабри обычно появляются в детстве. Первыми клиническими симптомами являются парестезии, ангиокератомы, непереносимость жары, помутнение роговицы, не приводящее к нарушению зрения, и гастроинтестинальные расстройства.
Патоморфологическая основа болезни Фабри начинает формироваться еще во внутриутробном периоде. Имеются гистопатологические свидетельства накопления GL-3 в сплетениях клеток почечной, печеночной и мышечной тканей, а также в оболочке кишечника у пораженных плодов мужского пола. Мутовчатое помутнение роговицы было обнаружено у 22-недельного плода мужского пола.
После рождения симптомы заболевания у мужчин проявляются в более раннем возрасте и имеют большую распространенность, чем у женщин. Обзор данных реестра болезни Фабри показал, что средний возраст появления симптомов у мужчин составил 6 лет, а у женщин – 9.
Первое проявление болезни в виде эпизодов жгучих невропатических болей в руках и ногах, известных как акропарестезии, было отмечено у 59% лиц мужского пола в возрасте 7 лет и у 41% женщин, но в возрасте 9 лет. Вторым симптомом, чаще всего встречающимся в детском возрасте, были гастроинтестинальные расстройства, имевшие место у 27% пациентов, но начинавшиеся у мальчиков с 5 лет, а у девочек – с 9,5. В детском возрасте также распространены гипергидроз и непереносимость жары. Хотя эти симптомы и не опасны для жизни ребенка, они существенно ухудшают качество его жизни.
Наиболее серьезные осложнения болезни Фабри возникают в зрелом возрасте и проявляются почечной недостаточностью, прогрессирующей кардиомиопатией, аритмиями, болезнями клапанного аппарата сердца и цереброваскулярными осложнениями, такими как инсульты. Развитие осложнений приводит к преждевременной смерти пациентов.
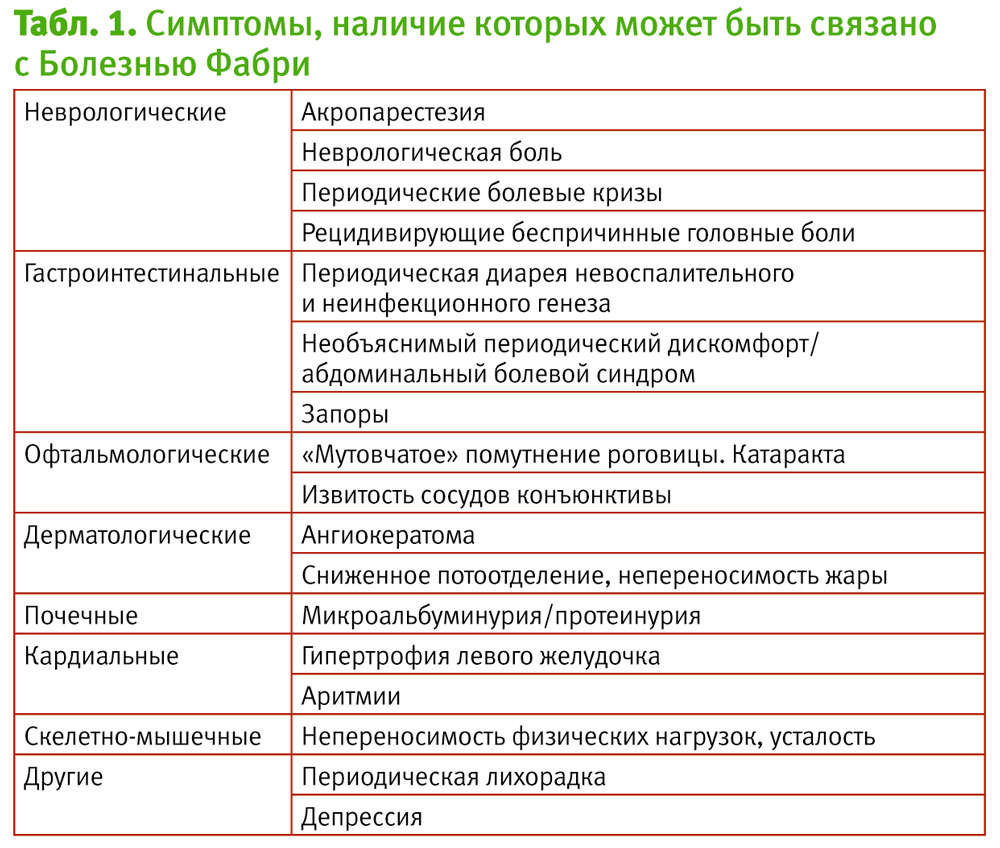
Неврологические
расстройства
Невропатическая боль, или акропарестезия, является первым симптомом заболевания и отмечается у 60–80% детей с классической болезнью Фабри. Пациенты описывают акропарестезии как длительные, интенсивные жгучие, колющие, изнуряющие боли, возникающие в дистальных отделах конечностей. Эти симптомы отражают повреждение мелких волокон в периферическом звене вегетативной нервной системы, возникающие в результате накопления гликосфинголипидов.
Боль – хроническая средней интенсивности, присутствует практически постоянно, эпизодически становясь практически нестерпимой (кризы Фабри). Кризы продолжаются от нескольких часов до нескольких суток.
Спровоцировать болевой криз может физическая нагрузка, усталость, стресс, гипертермия, пребывание в душном помещении, перемена погоды. Особенностью болевого синдрома при болезни Фабри является его устойчивость к действию обезболивающих препаратов.
В ряде случаев с возрастом у пациентов с болезнью Фабри интенсивность акропарестезий уменьшается в результате развивающихся снижений тактильной, температурной и болевой чувствительности. Однако большинство пациентов испытывают боль в течение всей жизни.
Невропатическая боль часто сопровождается ангидрозом или гипогидрозом (отсутствие или снижение потоотделения), что еще более ухудшает качество жизни пациентов и усугубляет непереносимость жары, душных помещений и физических нагрузок.

Ангидроз и гипогидроз у пациентов с болезнью Фабри становится причиной частых обморочных состояний.
В то же время, снижение потоотделения нельзя считать симптомом, патогномоничным для болезни Фабри, так как у части пациентов встречается гипергидроз (избыточное потоотделение).
Нарушение потоотделения возникает из-за отложения GL-3 в тканях потовых желез, сосудов, которые снабжают их кровью, а также из-за нарушения работы периферического звена вегетативной нервной системы. Нарушение потоотделения особенно характерно для детей и подростков.
У пациентов с поздними проявлениями болезни Фабри патология нервной системы проявляется преходящими нарушениями мозгового кровообращения и инсультами. В детской популяции эти клинические симптомы редки, хотя описаны случаи нарушения мозгового кровообращения у 12-летних пациентов.
Дерматологические
проявления
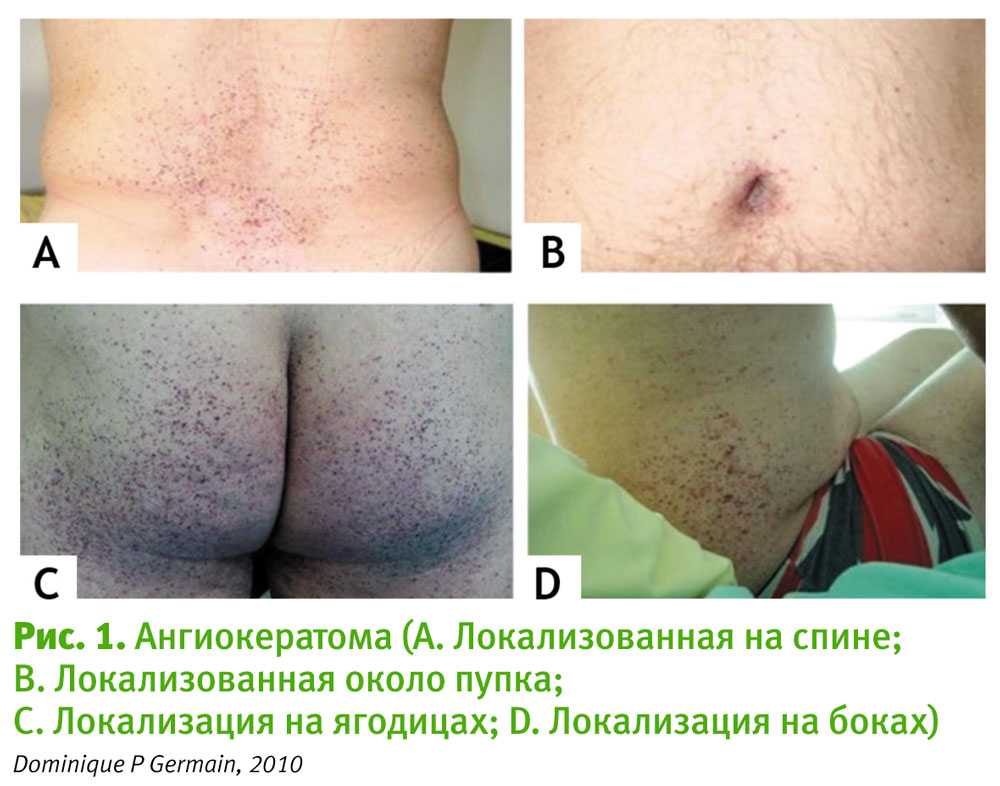
Типичные кожные проявления болезни Фабри – ангиокератомы – могут появляться у пациентов в подростковом возрасте. Ангиокератомы представляют собой мелкие, выступающие над поверхностью кожи безболезненные ангиомы темно-красного цвета, не меняющие цвет при надавливании. Первые элементы в большинстве случаев бывают единичными и могут иметь нетипичную локализацию (уши, грудная клетка). В дальнейшем ангиокератомы приобретают диффузный, иногда сливной характер, могут сливаться и располагаются в области бедер, пупка, ягодиц, нижней части живота и промежности (рис. 1), но иногда могут локализоваться на ладонях, вокруг рта, губ, а также на слизистых оболочках. Среди других кожных проявлений отмечаются лимфатические отеки нижних конечностей, изменения плотности волос на теле.
Поражение почек
Наиболее серьезным проявлением болезни Фабри является поражение почек, приводящее к формированию тяжелой почечной недостаточности и являющейся одной из основных причин летального исхода болезни Фабри. Причиной нарушения функции почек является накопление GL-3 в гломерулярном эпителии, клетках мезанглии, в интерстиции и подоцитах. Одним из первых признаков дисфункции почек, как правило, имеющей место у детей и подростков с болезнью Фабри, является микропротеинурия и микроальбуминурия (экскреция альбумина с мочой от 30 до 300 мг/сут). По мере развития заболевания формируется хроническая почечная недостаточность, требующая использования гемодиализа и в дальнейшем – проведения трансплантационных мероприятий.
Сердечно-сосудистая
система
Причиной кардиологических проявлений болезни Фабри становится накопление GL-3 практически во всех структурах сердца: эндокарде, миокарде, проводящей системе, крупных и мелких коронарных сосудах, вегетативной нервной системе, регулирующей сердечный ритм. При болезни Фабри, прежде всего, страдает проводящая система миокарда и клапанная система сердца, что в детском и подростковом возрасте проявляется периодическими подъемами артериального давления, как правило, бессимптомными вариантами аритмий, зарегистрированными на электрокардиограмме. В старшем возрасте пациенты могут предъявлять жалобы на боли в области сердца, сердцебиение, головокружение, диспноэ, синкопальные состояния.
Во время обследования пациента обнаруживается гипертрофия левого желудочка, изменения ЭКГ в виде расширения комплекса QRS, укорочение интервала PR, признаки атриовентрикулярной блокады и дисфункции синусового узла. Кардиологические осложнения – еще одна причина сокращения продолжительности жизни и раннего летального исхода у пациентов с болезнью Фабри.
Глазные симптомы
Самым характерным признаком болезни Фабри является «мутовчатое» помутнение роговицы (так называемая вортексная кератопатия, или cornea verticillata) – изменение в роговице, напоминающее пучок листьев или лепестков на конце стебля и, как правило, не приводящее к нарушениям зрения (рис. 2).
Кроме того, у пациентов регистрируется помутнение хрусталика в виде радиальной задней субкапсулярной катаракты – катаракты Фабри. Оба симптома являются патогномоничными и при обнаружении этих признаков можно с большой долей уверенности предположить наличие у пациента болезни Фабри. Другие, неспецифические проявления нарушений со стороны органа зрения – конъюнктивальные аневризмы, отек зрительной сетчатки и/или диска зрительного нерва, атрофия зрительного нерва, а также расширение и извитость сосудов глазного дна. У больных может развиваться оптический неврит, проявляющийся нарушениями полей зрения с формированием центральных скотом.
Гастроинтестинальные
симптомы
Гастроэнтерологические симптомы у пациентов с болезнью Фабри возникают достаточно рано, но в силу малой специфичности служат лишь косвенными маркерами болезни Фабри. Часто наблюдаются схваткообразные боли в животе, вздутие живота, неустойчивый стул, тошнота, рвота, снижение аппетита и дефицит веса.
Другие симптомы
заболевания
У 55% женщин и 39% мужчин отмечалось нарушение слуха и шум в ушах. К более редким и менее специфичным симптомам заболевания относятся особенности внешности (изменения по типу акромегалии), нарушения дыхания по типу ночного апноэ, анемия, скелетные аномалии, утолщение концевых фаланг пальцев по типу барабанных палочек), остеопороз, трещины и эритематозные изменения грибовидных сосочков на дорзальной поверхности языка, глоссит, гранулематозный хейлит, гипотиреоз, задержка полового развития, приапизм и др.
Диагностика
Для выявления болезни Фабри необходимы данные лабораторных методов диагностики, т. к. клиническая картина болезни разнообразна и не имеет типичных черт. Представителям мужского пола диагноз может быть подтвержден при обнаружении пониженной активности альфа-галактозидазы А в крови (лейкоциты), материале биопсии почек, культуре кожных фибробластов или других биологических средах. Уровень альфа-галактозидазы А в крови у девочек и женщин-носителей может не изменяться даже при наличии клинических проявлений болезни Фабри, что связано с инактивацией Х-хромосомы.
«Золотым стандартом» диагностики болезни Фабри и наиболее точным диагностическим методом является молекулярно-генетический анализ и обнаружение специфических (патогенетических) мутаций в гене GLA.
Если в семейном анамнезе беременной женщины есть данные о болезни Фабри, может быть проведена пренатальная диагностика – исследование ворсин хориона и/или культуры клеток амниотической жидкости на 9–11-й неделе беременности (определение активности альфа-галактозидазы А и проведение ДНК-анализа).
Лечение
С 2001 г. в мировой медицинской практике используют специфическую патогенетическую ферментозаместительную терапию болезни Фабри. Больные получают препарат Фабразим (b-агалсидаза), что позволяет не только замедлить течение этого тяжелого, неуклонно прогрессирующего заболевания, но и при своевременной диагностике и раннем начале лечения предотвратить отложение GL-3 в лизосомах и сохранить тем самым нормальными функции жизненно важных органов.
Несмотря на то, что мы накапливаем все больше знаний о редких наследственных заболеваниях – при постановке диагноза мы думаем о них в последнюю очередь, уменьшая тем самым шансы пациента на нормальную жизнь. Только раннее начало ферментозаместительной терапии может сохранить здоровье пациента.
Среди факторов, мешающих врачу установить диагноз – неспецифический, субъективный характер ранних симптомов и отсутствие осведомленности среди врачей. Пациенты часто осматриваются множеством специалистов в различных клинических областях и прежде, чем диагноз будет заподозрен, а затем подтвержден, диагностическая задержка может занять несколько лет, а к этому времени болезнь может привести к необратимым изменениям в организме человека.
Благодаря успехам генетики и современной медицинской науки, эффективное лечение наследственных болезней становится реальностью, уменьшает боль и страдания многих людей. Только раннее начало лечения может дать пациенту шанс на полноценную здоровую жизнь.
Необходимо, чтобы больные дети нашей страны наряду со своими сверстниками в развитых странах мира имели равный доступ к качественной современной медицинской помощи, основанной на лучших подходах к ведению пациентов с редкими болезнями.
коментарів