

С клинической точки зрения в пренатальном периоде целесообразно выделять пиелоэктазию (ПЭК) и гидронефроз. По нашим данным (Н. П. Веропотвелян и соавторы, 1994), частота пиелоэктазий, обнаруживаемых пренатально, составляет 21,7 на 1000 плодов. В качестве диагностического критерия ПЭК используются численные значения передне-заднего размера почечных лоханок при поперечном сканировании во II триместре беременности — свыше
5 мм, а в III триместре — свыше 7 мм. При расширении передне-заднего размера лоханки свыше 10 мм устанавливается диагноз гидронефроза. Частота гидронефроза составляет в среднем 2,5–2,8:1000 плодов.
В 74,6% случаев пиелоэктазии встречаются у плодов мужского пола. Выявлено также, что у женщин с ПЭК плода в анамнезе риск появления этой патологии при следующей беременности возрастает в 6,1 раза. Таким образом, можно говорить о «предрасположенности» к ПЭК, которая может возникать под действием генетических факторов или влиянием окружающей среды. Анализ частоты выявления ПЭК позволяет говорить о наследовании малых аномалий мочевых путей по аутосомно-доминантному признаку.
Для исключения дилатации почечной лоханки плода необходимо получать как поперечное, так и продольное сечение почек с оценкой их локализации, симметричности, формы, размеров, эхогенности паренхимы и состояния чашечно-лоханочного комплекса. О выраженности дилатации судят по степени увеличения передне-заднего размера лоханки, определяемом в поперечном сечении. Важно также исключить наличие мегауретера и оценить состояние мочевого пузыря и количество околоплодных вод.
Несмотря на существование большого количества классификаций, призванных определить степень тяжести поражения, применение их в практической медицине ограничено из-за отсутствия четких прогностических критериев и соответствующей пренатальной тактики. Однако нами (Н. П. Веропотвелян и соавторы, 1994) достаточно давно разработана и предложена классификация степени тяжести гидронефроза, которая реально применяется в СНГ:
- степень 0 — передне-задний размер лоханки <5 мм;
- степень I:
а) передне-задний размер лоханки 5–7 мм (прегидронефроз),
б) 8–12 мм (гидронефроз);
- степень II — передне-задний размер лоханки 13–15 мм, чашечки не затронуты;
- степень III — передне-задний размер лоханки >15 мм, чашечки дилатированы;
- степень IV — атрофия паренхимы.
По данным R. Wilson и соавторов, несмотря на то что пиелоэктазии в 3 раза чаще встречаются у плодов мужского пола, большинство из них носит «транзиторный» характер, в то время как у плодов женского пола вероятность сохранения патологических изменений в постнатальном периоде значительно выше. Выявлено также, что ПЭК сохраняются чаще при наличии одностороннего, чем двустороннего поражения, — 47% против 26%, при этом возрастает и вероятность применения хирургического лечения в постнатальном периоде.
Некоторые авторы рекомендуют проведение тщательного обследования новорожденных в случаях антенатальной регистрации передне-заднего размера лоханки >5 мм в любом гестационном сроке, другие исследователи считают, что при выявлении ПЭК >10 мм в любом сроке беременности нет необходимости в проведении комплексного обследования ребенка в постнатальном периоде.
В. Langer и соавторы делают вывод, что регистрация пиелоэктазии >5 мм до 28 недель сама по себе не имеет никакого прогностического значения без проведения динамического ультразвукового исследования, а любое расширение >10 мм, выявленное во II триместре беременности, нельзя считать патологическим без динамического контроля в III триместре.
Пиелоэктазию относят к «мягким» ультразвуковым признакам хромосомных аномалий (ХА). Суммируя результаты ряда исследований, R. Snijders и К. Nicolaides пришли к выводу, что общая частота встречаемости ХА у плодов с ПЭК составила 8%. При изолированном поражении этот показатель не превышал 2%, при наличии сочетанных ультразвуковых изменений был
во много раз выше — 33%.
Частота ХА при пиелоэктазии возрастает при наличии других эхографических изменений. В работе R. Snijders и К. Nicolaides была доказана зависимость частоты ХА от количества эхографических маркеров. В этом исследовании при наличии изолированной ПЭК вероятность выявления синдрома Дауна повышалась в 1,5 раза, при выявлении одной сочетанной аномалии — в 15 раз, двух — в 46 раз, трех и более — в 56 раз. Авторы считают, что пренатальное кариотипирование обосновано только в случаях сочетания ПЭК с другими эхографическими признаками патологии. Помимо синдрома Дауна, пиелоэктазия может встречаться у плодов с другими ХА. По данным литературы, у плодов с пренатально установленным диагнозом ХА, помимо трисомии 21, диагностируются синдромы Патау (37%), Эдвардса (18%), Тернера (8%), триплоидия (4%). Таким образом, учитывая наличие определенной связи между пиелоэктазией и ХА, вопрос о пренатальном кариотипировании следует решать в каждом случае индивидуально с учетом других факторов риска и эхографических изменений в других органах и системах.
Прогноз
Умеренно выраженная, изолированная пиелоэктазия имеет благоприятный прогноз. По данным педиатров, частота умеренно выраженной пиелоэктазии у детей от 2 месяцев до 13 лет составляет 4,8%. Чем раньше возникает ПЭК у плода, сохраняющаяся в течение всей беременности, тем хуже прогноз. Доказано также, что чем больше выражено расширение чашечно-лоханочной системы пренатально, тем больше страдает функция почки в постнатальном периоде. В любом случае пренатальное выявление расширения почечной лоханки требует обязательного динамического наблюдения, так как прогрессирование патологических изменений в III триместре беременности может привести к «отмиранию» почки и отсутствию ее функции в неонатальном периоде. Пренатальное исчезновение расширения почечной лоханки также не всегда является благоприятным признаком, так как уменьшение или исчезновение выраженного гидронефроза (35–50 мм) в начале III триместра может свидетельствовать о пренатальном сморщивании почки.
Тактика ведения
По данным М. В. Медведева, при обнаружении расширения передне-заднего размера почечной лоханки плода >10 мм во II триместре и >12 мм в III триместре беременности необходимо рекомендовать обследование ребенка в неонатальном периоде, так как в этой группе отмечается достоверно высокая частота сохранения патологических изменений и необходимости хирургического лечения.
Пренатальные диагностические критерии обструктивных уропатий верхних мочевых путей позволяют дифференциально определить транзиторные пиелоэктазии и достаточно рано отделить группу плодов с патологической дилатацией верхних мочевых путей, что дает возможность разработать наиболее рациональную тактику ведения. Пренатальная диагностика обструктивных уропатий верхних мочевых путей у плода позволяет не проводить прерывания беременности, а определить оптимальный объем специализированных мероприятий для новорожденного, которые включают консервативное лечение и хирургическую коррекцию.
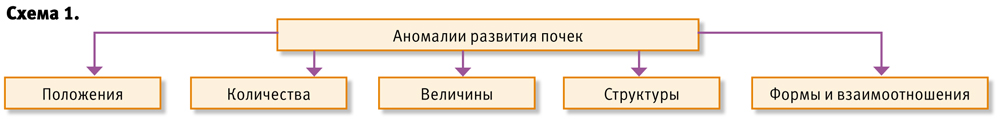
Аномалии развития почек: положения, количества, величины, структуры, формы, взаимоотношения (схема 1).
Аномалии величины почек: аплазия, гипоплазия, гипоплазия с дисплазией.
Агенезия почек плода (Поттер 0)
Почечная агенезия обусловлена или нарушением этапов эмбриогенеза при переходе от пронефроса к метанефросу, или недоразвитием мочеточникового зачатка. Она может быть двусторонней и односторонней.
Односторонняя агенезия означает отсутствие одной почки, обычно слева, и чаще носит семейный характер. Частота: 1 случай на 1000 новорожденных, но диагностируется пренатально не всегда. Это объясняется тем, что при одностороннем нарушении сохраняется нормальное количество околоплодных вод и визуализируется эхотень мочевого пузыря. Кроме того, изображение надпочечника или даже почечного ложа может быть ошибочно принято за изображение почки.
Косвенным признаком, указывающим на одностороннюю агенезию, может служить компенсаторное увеличение контралатеральной почки. При доплерометрии почечная артерия со стороны поражения не визуализируется. Из сочетанных аномалий чаще всего сопутствуют другие аномалии мочеполовой системы (агенезия надпочечников, у девочек — аномалии влагалища, у мальчиков — придатков яичка и семявыводящего протока), пороки желудочно-кишечного тракта (чаще других встречается неперфорированный анус), нарушения развития поясничного отдела позвоночника, а также синдромальная патология (например, VATER-ассоциация: Vertebral anomalies — нарушение строения позвонков, Anal atresia — атрезия ануса, Tracheo Esophageal fistula — трахеопищеводный свищ, Radial and/or Renal anomalies — аномалии лучевых структур и/или почек).
Прогноз при односторонней почечной агенезии благоприятный, однако риск развития почечной патологии, инфекций мочевыводящей системы и развития хронической почечной недостаточности и других урологических проблем выше, чем в популяции.
Двусторонняя агенезия почек (ДАП) означает полное отсутствие почек, мочеточников и относится к наиболее выраженным аномалиям МВС. Частота: в среднем 1 случай на 3000–5000 родов. У мальчиков в два раза чаще, чем у девочек. ДАП либо представлена спорадическими случаями, либо входит в состав синдромов с различным типом наследования (VATER, Fraser). Тератогенные факторы — сахарный диабет, краснуха, кокаин, щелочи.
На эхограммах характерна триада признаков:
- маловодие (может обнаруживаться после 16–18 недель);
- отсутствие эхотени мочевого пузыря;
- отсутствие изображения почек.
Однако пренатальная ультразвуковая диагностика ДАП может быть сильно затруднена в связи со сходной эхографической картиной с другими выраженными двусторонними изменениями почек (аплазия, гипоплазия, дисплазия). По данным A. Reuss и соавторов и J. Scott и М. Renwick, точность пренатальной диагностики ДАП составляет 69–73%. Важным дополнительным критерием двусторонней агенезии почек является отсутствие изображения почечных артерий в режиме цветового доплеровского картирования (ЦДК). Точность диагностики ДАП при использовании только В-режима составляет 80%, в режиме ЦДК — 100%. Однако ЦДК не всегда может помочь в установлении окончательного диагноза ДАП, так как почечные артерии могут иметь резко суженый просвет и не визуализироваться при диспластичных и гипоплазированных почках.
ДАП часто сопутствует задержка внутриутробного развития плода (ЗВРП) и фенотип Поттер, который относится к последствиям маловодия и включает:
- гипоплазию легких;
- специфические черты лица (эпикант, приплюснутый нос, срезанный подбородок, крыловидные кожные складки, низко расположенные уши);
- деформации верхних и нижних конечностей (искривление костей, косолапость, врожденные вывихи бедра).
Сочетанные пороки при ДАП присутствуют практически всегда. ДАП описана более чем при 140 синдромах с множественными пороками развития.
Прогноз при двусторонней агенезии почек летальный (гипоплазия легких, отсутствие деятельности МВС с прогрессирующей уремией, сочетанные пороки и ЗВРП). Поэтому при обнаружении ДАП целесообразно рекомендовать прерывание беременности на любом сроке. Предварительно необходимо провести кариотипирование для исключения ХА. Обязательным должно являться патологоанатомическое исследование для исключения синдромальной патологии.
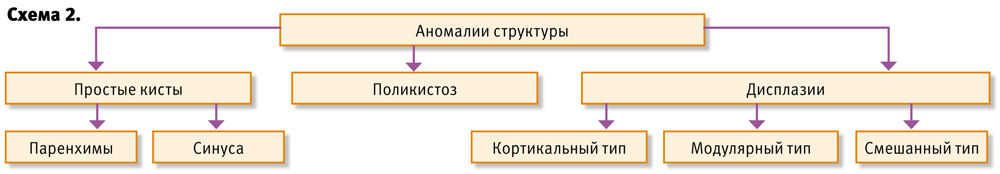
Аномалии структуры, в основе — различные виды кистозных изменений почечной ткани, которые являются следствием нарушения эмбриогенеза, представлены на схеме 2).
Аутосомно-рецессивная поликистозная болезнь почек (Поттер I)
При аутосомно-рецессивной поликистозной болезни почек (АРПБП) инфантильного типа отмечается вторичная дилатация и гиперплазия нормально сформированных собирательных канальцев почек вследствие несвоевременного и неправильного со-
единения секреторного и экскреторного аппаратов почки. Почки поражаются симметрично, мелкими кистами размерами 1–2 мм. Кисты через расширенные канальцы чаще сообщаются с лоханкой («открытые» кисты), способны выделять мочу и обеспечивать функцию почек. Если кисты «закрытые», то плод (или ребенок в первые дни жизни) погибает. Частота: 1 случай на 4000 родов, у плодов женского пола встречаются чаще.
Риск повторения заболевания у одного из детей составляет 25%. Ген, ответственный за АРПБП, располагается в коротком плече 6-й хромосомы. Поэтому целесообразно проведение пренатального кариотипирования и определение кариотипа родителей. Другие формы поликистозных почек могут иметь схожую эхографическую картину, но не сопровождаются специфичным генным нарушением. В случаях смерти плода или новорожденного необходимо проведение полного патологоанатомического исследования.
Основные пренатальные эхографические критерии АРПБП во второй половине беременности: значительно и симметрично увеличенные гиперэхогенные почки, отсутствие эхотени мочевого пузыря и маловодие, кисты из-за малого их размера не визуализируются, степень васкуляризации паренхимы может быть не изменена, что является относительно благоприятным прогностическим признаком. Иногда увеличенные почки занимают большую часть поперечного сечения живота плода. Некоторые исследователи приводят данные, что типичная эхографическая картина может не появляться до III триместра беременности.
Прогноз при аутосомно-рецессив-ной поликистозной болезни почек в случаях раннего проявления заболевания неблагоприятный. При последующих беременностях рекомендуется проведение аспирации ворсин хориона в 11–12 недель для исключения мутаций локуса 6р21.
Аутосомно-доминантная поликистозная болезнь почек (Поттер III)
Аутосомно-доминантная поликистозная болезнь почек (АДПБП) наследуется по аутосомно-доминантному типу. Кистозному перерождению подвергаются и нефрон, и собирательные канальцы. Приблизительно 1 человек из 1000 является носителем гена АДПБП в общей популяции.
Эхографические признаки АДПБП в пренатальном периоде появляются редко (множественные кисты в паренхиме почки начинают выявляться после 7-летнего возраста). Поэтому большинство случаев заболевания пренатально не диагностируется. В некоторых случаях к концу II и в III триместре беременности определяется двусторонняя умеренно выраженная нефромегалия и кисты различных размеров. Паренхима почек может быть гиперэхогенной; количество околоплодных вод обычно нормальное или слегка уменьшенное; мочевой пузырь, как правило, визуализируется. Среди сочетанных аномалий при АДПБП могут быть аномалии клапанов сердца, внутримозговых сосудов, печени.
Дифференциальная диагностика при аутосомно-доминантной поликистозной болезни почек проводится с мультикистозной дисплазией и другими видами почечных дисплазий, с новообразованиями почек, с синдромами гиперроста, а также с проявлениями врожденного нефротического синдрома и внутриутробного инфицирования. Для АДПБП характерно наличие семейного «почечного» анамнеза: кисты почек у родственников, пиелонефриты, проявления хронической почечной недостаточности.
При определении прогноза при аутосомно-доминантной поликистозной болезни почек ключевым фактором является количество околоплодных вод. При нормальном количестве околоплодных вод прогноз для жизни относительно благоприятный. Однако впоследствии могут развиться гипертензия и хроническая почечная недостаточность разной степени тяжести. При маловодии прогноз неблагоприятный.
Риск повтора плодовой формы при следующей беременности составляет 50%, особенно при наличии почечной патологии у матери или отягощенной наследственности по материнской линии. Поэтому при выявлении признаков АДПБП у плода необходимо тщательное ультразвуковое исследование почек родителей, изучение семейного анамнеза и решение вопроса о зондовой генетической пренатальной диагностике.
Мультикистозная дисплазия почек (Поттер II)
Мультикистозная дисплазия почек (МДП) относится к группе нарушений раннего этапа эмбрионального развития почек. Причиной кистозного перерождения почечной ткани является отсутствие закладки экскреторного при сохранении секреторного аппарата почки, и как следствие — ранние обструкции мочевыводящих путей. Чаще поражается одна почка. МДП возникает обычно спорадически, реже может входить в состав синдромов МВПР.
Частота: 1 случай на 3000–5000 родов при одностороннем поражении и
1 случай на 10000 родов при двустороннем поражении. По данным D. Wellesley и D. Howe, односторонние поражения составляют 76% всех случаев МДП, двусторонние — 24%. Соотношение встречаемости заболевания у мальчиков и девочек составляет 2,4:1.
Эхографическая картина мультикистозной дисплазии почек в пренатальном периоде появляется уже в начале II триместра беременности: одна почка без особенностей, на стороне поражения определяется выраженная нефромегалия за счет множественных крупных тонкостенных кист, которые не соединяются между собой (степень увеличения зависит от размеров кист, размеры которых могут варьировать от 1 до 10 см). Паренхима почек между кистами гиперэхогенная, почка имеет неровные контуры. Количество околоплодных вод и мочевой пузырь при одностороннем поражении не изменены и резко уменьшены при двустороннем поражении. При доплерометрии почечных сосудов на стороне поражения почечная артерия может быть резко суженной, с турбулентным характером кровотока, отсутствием диастолического компонента и редукцией систолического пика или не определяться вообще.
Точность пренатальной ультразвуковой диагностики МДП составляет 95%. Среди сочетанных пороков наиболее часто отмечаются пороки МВС, в 25% случаев имеется различная патология второй почки. МДП может входить в состав множественных пороков, среди которых преобладают аномалии сердца, позвоночника, конечностей, лица и пуповины. МДП может сочетаться с хромосомными аномалиями и входить в состав более 50 синдромов с различным типом наследования. Односторонние и изолированные случаи МДП редко сопровождаются ХА, а двусторонние — втрое чаще сочетаются с экстраренальной патологией и нарушениями кариотипа.
Дифференциальная диагностика при МДП проводится с другими типами кистозных заболеваний почек (аутосомно-рецессивная поликистозная болезнь, аутосомно-доминантная поликистозная болезнь), с другими видами почечных дисплазий, с проявлениями обструктивных уропатий, с обструктивными пороками желудочно-кишечного тракта, а также с новообразованиями почек, проявлениями врожденного нефротического синдрома и внутриутробного инфицирования, то есть состояний, при которых могут визуализироваться гиперэхогенные почки, содержащие кисты.
Прогноз при мультикистозной дисплазии почек зависит главным образом от формы поражения: одно- или двустороннее, а также наличия синдромальной патологии и сочетанных пороков.
При односторонней форме заболевания прогноз для жизни благоприятный. Однако ребенку может потребоваться хирургическое лечение, и остается повышенный риск развития хронической почечной недостаточности, увеличивающийся с возрастом.
При двусторонней форме заболевания с выраженными пренатальными проявлениями прогноз неблагоприятный. Двусторонняя МДП сопровождается повышенным риском ХА и сочетанных аномалий.
Мультилокулярная киста — многокамерная киста почки, заключенная в капсулу, с собирательной системой не сообщается. Локализуется в одном из полюсов почек. На эхограммах: одна почка не изменена, вторая увеличена в размере, контур неровный за счет выбухания в области полюса из-за объемного образования, содержащего множественные не соединяющиеся между собой мелкие кисты, разделенные гиперэхогенными перегородками. Чашечно-лоханочная система и оставшаяся паренхима не изменены.
Медуллярная киста — врожденное перерождение дистальных отделов мальпигиевых пирамид (кистозное поражение канальцевого аппарата). Процесс двусторонний, но в него может вовлекаться только часть почки. На эхограммах: размер почек в норме, контур ровный, корковый слой паренхимы без особенностей, мозговой слой — пирамидки гиперэхогенные, с наличием в них мелких кист, невидимых на эхограммах, создающих большое количество отражающих поверхностей, в связи с чем пренатальная идентификация медуллярных кист затруднена.
Одиночные кисты почек плода
Солитарные кисты — инфантильные кисты почек, как правило, бывают представлены округлой или овальной формы анэхогенными образованиями с четким ровным контуром различной локализации, чаще однокамерными, чашечно-лоханочная система не изменена.
Их стенка состоит из однослойного эпителия, а содержимым является чистая серозная жидкость. Эти кисты не имеют сообщения с почечной лоханкой. Размеры почек находятся в пределах нормативных значений.
Частота простых кист почек у детей первого года жизни составляет в среднем 16:10000. Соотношение встречаемости кист у мальчиков и девочек, так же как и между правой и левой почками, приблизительно одинаковое. Чаще всего кисты обнаруживаются в верхнем полюсе почки. Этиология инфантильных кист почек остается не установленной.
В пренатальном периоде инфантильные кисты почек обычно выявляются после 20–22 недели беременности в виде односторонних однокамерных округлой формы анэхогенных образований. Размеры кист варьируют от 3 до 70 мм, составляя в среднем 10 мм.
Прогноз при одиночных простых кистах почек благоприятный. Хотя результатов длительного наблюдения за детьми с инфантильными кистами почек опубликовано не много, но вывод этих работ однозначный — размеры кист не увеличиваются и они не вызывают нарушений функции почек.
Продолжение статьи читайте
в следующем номере.
коментарів