

Класифікація МКХ 10: Е 21.
Визначення
Гіперпаратиреоз – гетерогенна група станів, що характеризується гіперфункцією прищитоподібних залоз (ПЩЗ).
Патогенетично виділяють: первинний, вторинний і третинний гіперпаратиреоз.
В основі первинного гіперпаратиреозу лежить гіперплазія або аденома ПЩЗ. Він характеризується поєднанням високої концентрації паратгормону (ПТГ) з гіперкальціємією. Вторинний гіперпаратиреоз – нормальна реакція ПЩЗ на тривалу гіпокальціємію у зв'язку дефіцитом вітаміну D або із захворюваннями, що вражають нирки, печінку, кишківник. Вторинний гіперпаратиреоз проявляється високою концентрацією ПТГ і гіпокальціємією. Третинний гіперпаратиреоз виникає у пацієнтів з давнім вторинним гіперпаратиреозом і супроводжується підвищеною концентрацією ПТГ в сироватці крові та появою гіперкальціємії.
Первинний гіперпаратиреоз (Е 20.0)
Це синдром, що обумовлений надлишковою продукцією ПТГ за рахунок ураження безпосередньо прищитоподібних залоз, наприклад гіперплазії або аденоми, та супроводжується гиперкальціємією.
Це третя за розповсюдженістю ендокринологічна патологія, частота якої варіює від 0,5 до 34 на 1000 населення у залежності від методів виявлення.
Первинний гіперпаратиреоз в середньому діагностується з частотою 25 нових випадків на 100 тис. населення на рік. З ним пов'язано 20% випадків синдрому гіперкальціємії.
Переважна більшість аденом ПЩЗ (80% випадків), з якими і пов'язують розвиток первинного гіперпаратиреозу, розташовуються у типовому місці – за щитоподібною залозою.
У дітей гіперпаратиреоз зустрічається рідко і виникає, як правило, внаслідок розвитку доброякісної аденоми.
Зазвичай клінічні прояви з'являються після 10-річного віку.
Етіологія
Основні причини первинного гіперпаратиреозу:
1) солітарна (80%) або множинні (5%) аденоми ПЩЗ;
2) гіперплазія ПЩЗ (15%);
3) карцинома ПЩЗ (<5%);
4) первинний гіперпаратиреоз у межах синдромів множинних ендокринних неоплазій 1-го та 2-го типу (МЕН-1, МЕН-2).
Патогенез
Підвищена продукція паратгормону призводить до надмірного виведення нирками фосфатів. Зниження сироваткового рівня останніх стимулює синтез 1,25-(OH)2-D3, який сприяє всмоктуванню надлишку Ca2+ у кишківнику. Далі гіперкальціємія підсилюється за рахунок активації остеокластів надлишком паратгормону. Збільшена кількість паратгормону призводить до прискорення обміну в кістках, прискорення кісткової резорбції та кісткового утворення. Але формування нової кісткової тканини відстає від її розсмоктування, що викликає генералізований остеопороз та остеодистрофію, вимивання кальцію з кісткових депо та гіперкальцемію. Також виникає кальціурія, наслідком якої є пошкодження епітелію ниркових канальців, а також утворення каменів у нирках. Нефрокальціноз, в свою чергу, веде до порушення функцій нирок. Підвищення рівнів концентрації кальцію і фосфатів у сироватці крові можуть привести до кальцифікації судин кровоносного русла, обумовлюючи ризик передчасної смерті.
Клініка
Основними скаргами хворих є стомлюваність, м'язова слабкість, біль у кістках, зниження апетиту, нудота, блювота. Більш ніж в 50% випадків діагноз встановлюється при випадковому виявленні гіперкальціємії. Клінічні прояви хвороби пов'язані з рівнем гіперкальціємії. При рівні концентрації кальцію у сироватці крові 3,0–3,5 ммоль/л виникають нездужання і сонливість, 3,5–4 ммоль/л — можливий розвиток психозу, а при концентрації кальцію в сироватці крові понад 4 ммоль/л спостерігається порушення свідомості, розвиток коми і можливий летальний результат.
Симптоматика первинного гіперпаратиреозу складається з наступних груп симптомів:
1) Ниркова симптоматика (40–50%). Нефролітіаз, рідше – прогностично несприятливий нефрокальціноз. Рефрактерний до дії антидіуретичного гормону інсипідарний синдром (поліурія, полідипсія, гіпоізостенурія), який у важких випадках призводить до ниркової недостатності.
2) Кісткова симптоматика (50%). Гіперпродукція паратгормону призводить до негативного кісткового балансу. Рентгенологічно виявляється дифузна остеопенія: при дослідженні кистей – у 40% випадків, хребців – у 20%. У важких випадках може встановлюватися патогномонічна субперіостальна резорбція і акроостеоліз кінцевих фаланг кистей і стоп. Кісти, гігантськоклітинні пухлини і епуліди у даний час виявляються винятково рідко.
3) Гастроінтестинальна симптоматика (50%). Анорексія, нудота, обстипація, метеоризм, схуднення, у 10% випадків мають місце пептичні виразки шлунку та/або дванадцятипалої кишки, у 10% – панкреатит (при загостренні рівень Са2+ може знижуватися), рідше панкреакалькульоз. Удвічі частіше, ніж у популяції, зустрічається жовчнокам'яна хвороба.
4) Нейром'язова симптоматика. М'язова слабкість і атрофія, скорочення інтервалу QT на ЕКГ. Депресія, сонливість, ослаблення пам'яті.
5) Ураження серцево-судинної системи при гіперпаратиреозі характеризується артеріальною гіпертензією, гіпертрофією лівого шлуночка, кальцифікацією коронарних артерій та клапанів серця.
6) Гіперкальціємічний криз – важке, загрозливе для життя ускладнення, що зустрічається менш ніж у 5% пацієнтів. Провокується гіподинамією, переломами кісток, призначенням тіазидних діуретиків. Клінічно гіперкальціємічний криз проявляється поліурією, полідипсією, блювотою, ексикозом, адинамією, сомноленцією, комою.
Первинний гіперпаратиреоз у новонароджених
Тяжкий первинний гіперпаратиреоз новонароджених – рідкісний розлад. В більшості пацієнтів є мутації гена CASR. Симптоми розвиваються незабаром після народження. Основними проявами є анорексія, дратівливість, млявість, закріпи, недостатня прибавка маси тіла. У новонароджених із важким гіперпаратиреозом спостерігається помітне підвищення кальцію і ПТГ у сироватці крові. Концентрація ПТГ дуже висока, а гіперкальціємія небезпечна для життя. Рентгенологічно визначається підокістна резорбція кістки, остеопороз, патологічні переломи. Гістологічно визначається дифузна гіперплазія прищитоподібних залоз. Зазвичай первинний гіперпаратиреоз новонароджених є спадковим ураженням. Діти з важким гіперпаратиреозом новонароджених можуть бути гомозиготними або гетерозиготними за мутацією гена кальцієвих каналів, у той самий час більшість людей з цією мутацією мають аутосомно-домінантну сімейну гіперкальціємію.
Множинні ендокринні неоплазії
МЕН I типу є аутосомно-домінантним захворюванням, яке характеризується гіперплазією або неоплазією ендокринної частини підшлункової залози (що виділяє гастрин, інсулін, панкреатичний поліпептид, а іноді і глюкагон), передньої долі гіпофіза (що зазвичай виділяє пролактин) і прищитоподібних залоз. У більшості випадків МЕН I зустрічається після 50-річного віку, і лише в рідкісних випадках у дітей до 18 років. Гіперпаратиреоз діагностується у 90% пацієнтів. У більшості хворих відзначається безсимптомна гіперкальціємія, в 25% випадків спостерігаються ознаки сечокам'яної хвороби. Наявність відповідних ДНК-зондів дозволяє виявити носіїв гена з 99% точністю при народженні, уникаючи непотрібних біохімічних скринінгових програм.
МЕН I виникає при мутації гена MEN1, який розташований на хромосомі 11q13, і кодує протеїн менін, що функціонує як супресор пухлин. Запропоновано гіпотезу двосторонньої рецесивної мутації, згідно з якою перша з них в домінантному алелі створює схильність до туморогенезу, а друга супроводжується елімінацією нормального алеля і демаскує дефектний алель, індукуючи розвиток пухлини.
Синдром МЕН IIа типу характеризується наявністю медулярної карциноми щитоподібної залози, феохромоцитоми (одиничної, білатеральної або множинної) і гіперпаратиреозу. Синдром був описаний Джоном Сіпплом у 1959 році. Він успадковується за аутосомно-домінантним типом з високим ступенем пенетрантності та експресивністю, що варіює.
В основі МЕН IIа лежать мутації у гені RET. Цей ген кодує білок, який бере участь у передачі сигналів у клітинах. Мутації у гені RET провокують зростання і поділ клітин за відсутності сигналів ззовні клітини. Це може привести до утворення пухлин в ендокринних залозах та інших тканинах.
Сімейна гіпокальціурична гіперкальціємія
Характеризується аутосомно-домінантним типом успадкування, нормальним рівнем ПТГ, помірною гіперкальціємією і гіпокальціурією. В основі захворювання лежать мутації гена CASR. Захворювання зазвичай протікає безсимптомно або проявляється неспецифічними симптомами у вигляді втоми, слабкості, артралгій, краніалгій.
Вторинний гіперпаратиреоз (Е 21.1)
Визначення
Вторинний гіперпаратиреоз – компенсаторна гіперфункція і гіперплазія прищитовидної залози, що розвивається при тривалій гіпокальціємії і гіперфосфатемії різного генезу.
Цей варіант гіперпаратиреозу найчастіше виникає у дитячому віці.
Етіологія
Причинами вторинного гіперпаратиреозу може бути:
- Ниркова патологія: хронічна ниркова недостатність, тубулопатії, нирковий рахіт.
- Кишкова патологія: синдром мальабсорбції.
- Кісткова патологія: остеомаляція, хвороба Педжета.
- Недостатність вітаміну D: захворювання нирок, захворювання печінки, спадкові ферментопатії.
- Злоякісні захворювання: мієломна хвороба.
Основними причинами вторинного гіперпаратиреозу є ниркова недостатність і хвороби системи травлення.
Патогенез
Гіперфосфатемія, обумовлена різними патофізіологічними факторами, зокрема, зменшенням маси функціонуючих нефронів, стимулює продукцію фактора росту фібробластів 23 (FGF-23) остеоцитам.
Свою дію FGF-23 реалізує через рецептор FGFR1, який функціонує лише у тому випадку, якщо він ко-експресований з трансмембранним протеїном Klotho у вигляді Klotho-FGF- рецепторного комплексу. У проксимальних канальцях нирок FGF-23, інгібуючи експресію натрій-фосфатного ко-транспортеров II типу (NaPi-2a і NaPi-2c), знижує реабсорбцію фосфатів. Тобто, FGF-23 до певного моменту підтримує нормальний рівень фосфатів у сироватці крові. Також FGF-23 зменшує синтез ПТГ. Однак FGF 23 інгібує активність 1-альфа-гідроксилази, опосередковуючи зниження рівня активного метаболіту вітаміну Д – кальцитріолу (1,25 (OH) 2D3). Зниження рівня кальцитріолу супроводжується дефіцитом порушення його рецептора (vitamin D receptor – VDR) у клітинах слизової оболонки кишечника, що призводить до зниження синтезу кальцій-зв'язуючих протеїнів, і в клітинах ПЩЗ, збільшуючи синтез мРНК ПТГ.
Дефіцит кальцітріолу зменшує всмоктування кальцію у кишечнику, що призводить до гіпокальціємії та розвитку остеомаляції. Гіпокальціємія стимулює продукцію ПТГ, що сприяє підсиленій кістковій реабсорбції та руйнуванню кістки. Гіперфосфатемія і збільшення продукції FGF-23 супроводжується зниженням експресії VDR і кальцій-чутливих рецепторів (calcium-sensing receptor – CaSR), у зв'язку з чим клітини ПЩЗ втрачають здатність адекватно реагувати на зміни концентрації кальцію та/або кальцитріолу.
При тривалому впливі високих концентрацій FGF-23 відбувається фосфорилювання екстрацелюлярної регульованої кінази 1/2 у клітинах ПЩЗ, що призводить до значного зниження експресії FGFR1 і білка Klotho. Зниження експресії Klotho-FGF-рецепторного комплексу розгальмовує продукцію ПТГ.
Таким чином, на ранніх стадіях розвитку вторинного гіперпаратиреозу гіперпродукція ПТГ переважно зумовлена порушенням вітамін Д-асоційованої регуляції обміну кальцію і функціонування CaSR, а в подальшому дефіцитом активності FGF-23-асоційованих сигнальних шляхів.
Тривала стимуляція ПТГ веде до гіперплазії ПЩЗ: спочатку до поліклональної гіперплазії, а потім до моноклональної вузлової гіперплазії. Ключовим фактором, який визначає проліферацію клітин і розвиток гіперплазії ПЩЗ, є зниження активності сигналів, асоційованих з CaSR. Зниження активності CaSR призводить до дефіциту активації фактора транскрипції Sp1 і, як наслідок, до дефіциту трансактивності VDR. Недостатність експресії CaSR і VDR обумовлює зниження чутливості клітин ПЩЗ до інгібуючого впливу кальцію і кальцитріолу на секрецію ПТГ і сприяє розвитку гіперплазії ПЩЗ.
Клініка
У клінічній картині вторинного гіперпаратиреозу домінують симптоми основного захворювання, частіше за все, хронічної ниркової недостатності. Специфічними симптомами гіперпаратиреозу є болі в кістках, слабкість у проксимальних відділах м'язів, артралгії. Можуть виникати спонтанні переломи і деформація скелета. Утворення позакісткових кальцинатів має різні клінічні прояви. При кальцифікації артерій можуть розвиватися ішемічні зміни. На руках і ногах можуть бути виявлені периартикулярні кальцинати. Кальцифікація кон'юнктиви і рогівки у сполученні з рецидивуючим кон'юнктивітом позначається як синдром «червоного ока».
Діагностика гіперпаратиреозу
Діагностика первинного гіперпаратиреозу ґрунтується на виявленні гіперкальціємії, зниженого рівня неорганічного фосфору, підвищеного рівня ПТГ в плазмі крові. Для діагностики первинного гіперпаратиреозу рекомендується проведення 3-кратного дослідження загального або іонізованого кальцію (більш інформативно) у сироватці крові з інтервалом 2–3 тижні. При первинному гіперпаратиреозі рівень кальцію підвищений, зазвичай >2,85 ммоль/л. Гіперкальціємія є більш вираженою у дітей з гіперплазією прищитоподібних залоз, але показники кальцію не перевищують значення 3,0 ммоль/л. Кількість іонізованого кальцію завжди значно збільшена, навіть якщо загальний рівень кальцію є граничним або злегка підвищеним. Рівень сироваткового фосфору і магнію значно зменшується. У сироватці крові підвищується рівень залишкового азоту та сечової кислоти. Рівень лужної фосфатази підвищується у 1,5–6 разів, але у дітей може бути нормальним. Сироватковий рівень ПТГ підвищений, корелює з рівнем кальцію, у той же час рівень концентрації кальцитоніну відповідає віковій нормі. Сеча може мати постійну низьку питому вагу (гіпоізостенурія).
Для вторинного гіперпаратиреозу характерні нормокальціємія або помірна гіпокальціємія в поєднанні з підвищеним рівнем паратгормону. Крім того, характерна гіперфосфатемія, високий рівень лужної фосфатази, низький рівень кальцитріолу. Визначення рівня паратгормону рекомендується при нефропатії будь-якого генезу зі зниженням швидкості клубочкової фільтрації менше 60%.
Для визначення об'ємних утворень або гіперплазії ПЩЗ використовують різні методи візуалізації: УЗД, сцинтіграфію, мультиспіральну комп'ютерну томографію (КТ). При малій масі ПЩЗ УЗД є малоінформативною процедурою. Більш ефективним методом візуалізації ПЩЗ є мультиспіральна КТ, яка дозволяє виявити аденоми розміром 0,2–0,3 см та їх атипову локалізацію. Для більш детальної топічної діагностики можливого розташування ОЩЖ проводиться сцинтиграфія з використанням технетріла 99mTc. Чутливість даного методу досягає 91%.
Для діагностики наявності остеопорозу проводять остеоденситометрію та рентгенологічне дослідження. Інформативною є остеоденситометрія поперекового відділу хребта, проксимального відділу стегнової, а також дистального відділу променевої кістки. Рентгенологічними ознаками первинного гіперпаратиреозу є дифузний остеопороз, витончення кортикального шару з розширенням кісково-мозкового каналу. Специфічною ознакою первинного гіперпаратиреозу є зменшення щільності кортикальної тканини в порівнянні з губчастою. Близько 10% хворих мають рентгенологічні ознаки рахіту. Кісткові зміни при вторинному гіперпаратиреозі схожі з первинним гіперпаратиреозом (остеопороз, субперіостальна і субхондральна резорбція кісток кисті та ін.).
Рентгенограма черевної порожнини може виявляти ниркові камені або нефрокальциноз. Наявність остеопорозу є показанням для дослідження рівнів маркерів кісткової резорбції і кісткоутворення. Маркерами кісткової резорбції є N- і C-телопептиди молекул колагену 1-го типу (NTX, CTX), тартратрезистентна кисла фосфатаза (TRACP), а маркерами кісткоутворення – остеокальцин, карбокси- і амінотермінальні про-пептиди проколагену 1-го типу (Р1СР, Р1NP), загальна лужна фосфатаза (ALP) та її кістковий ізофермент (bALP).
При вторинному гіперпаратиреозі необхідна діагностика основного захворювання (хронічна ниркова недостатність, мальабсорбція та ін.).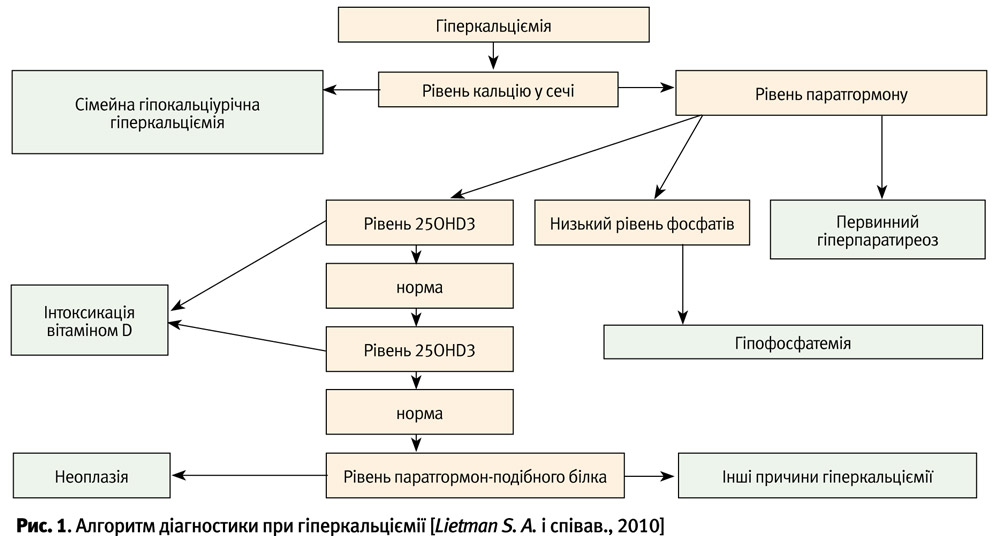
Диференційна діагностика первинного гіперпаратиреозу
Диференційна діагностика первинного гіперпаратиреозу проводиться із захворюваннями, що супроводжуються гіперкальціємією (рис. 1). Гіперкальціємія у дітей може спостерігатися при досить багатьох захворюваннях (табл. 1).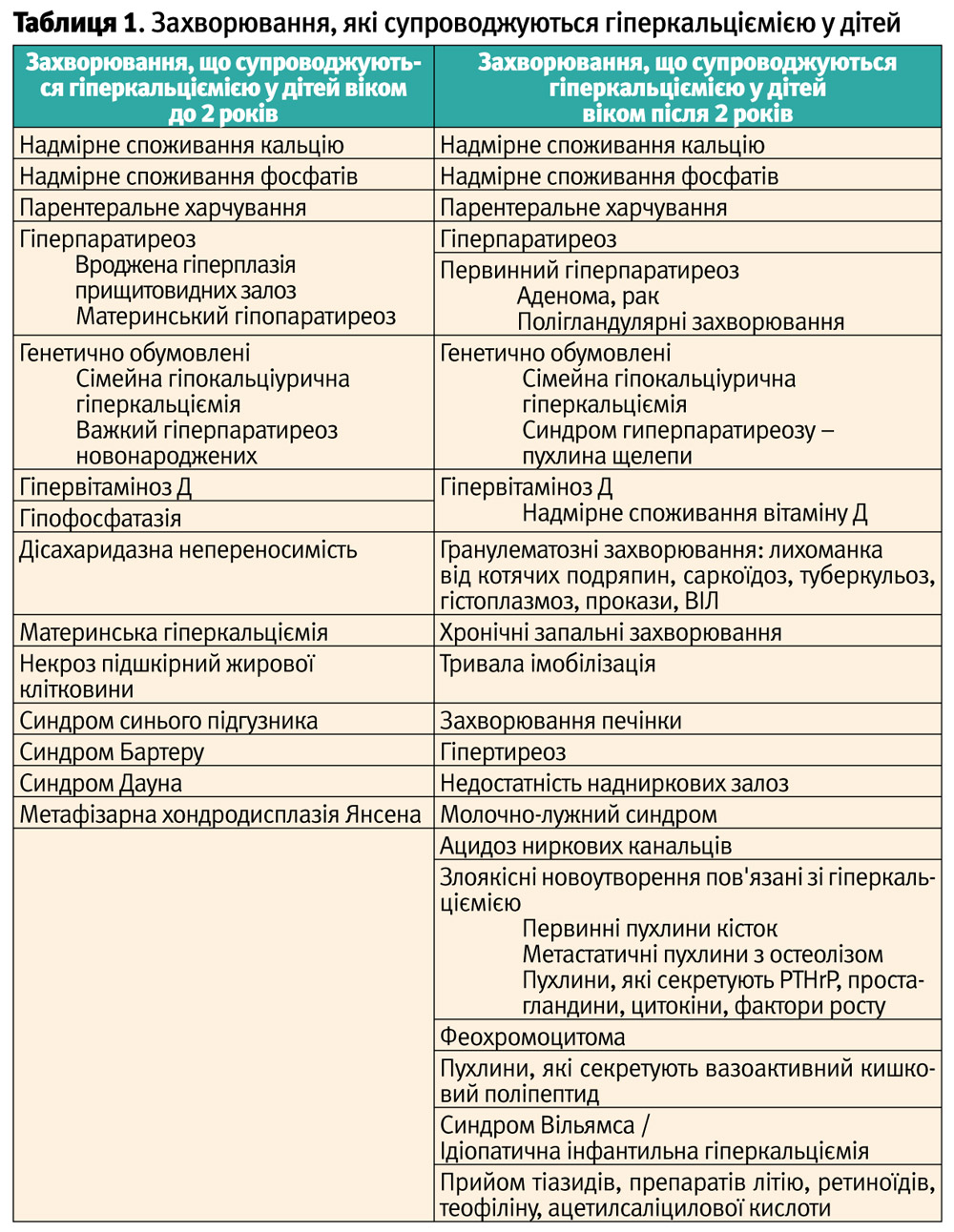
Лікування гіперпаратиреозу
При первинному гіперпаратиреозі практично у всіх випадках рекомендується оперативне лікування. Необхідне ретельне обстеження ПЩЗ. Якщо аденома виявлена, вона повинна бути вилучена. Більшість новонароджених з тяжкою гіперкальціємією потребують загальної паратиреоїдектомії, при легкій гіперкальціємії від хірургічного лікування можна утриматись. Після оперативного лікування пацієнт повинен отримувати лікування гіпопаратиреозу на протязі 6–12 місяців, потім рівні кальцію приходять до норми.
При вторинному гіперпаратиреозі показана профілактика остеопатії. З цією метою призначають активні препараті вітаміну D (альфа кальцидіол, кальцитріол). При триразовому підвищенні рівня паратгормону та/або підвищенні рівня кальцію крові більше 2,6–2,7 ммоль/л показана субтотальна паратиреоїдектомія.
Ефективність та безпека препаратів групи бісфосфонатів (алендронової, ібандронової, золедронової кислот) і кальціміметиків (цинакальцета) у дітей вивчені недостатньо.
Прогноз
Прогноз відносно сприятливий. Після успішного оперативного лікування більшість симптомів гіперпаратиреозу зникає протягом 6–12 місяців.
Перелік літератури знаходиться у редакції
коментариев