
Чи є ризик знову народити і чи можна уникнути повторного народження дитини, хворої на спадкові хвороби?
Для сімей, у яких є діти, хворі на спадкові хвороби, що мають рецесивний тип успадкування, ризик повторного народження такої ж хворої дитини становить 25% для кожної наступної вагітності. Такий показник вважається високим генетичним ризиком, оскільки у п’ять разів перевищує значення загально-популяційного ризику (5%) народження у пари дитини із вродженими та спадковими захворюваннями. Значення можливості народити здорову дитину для таких сімей неможливо перебільшити. Батьки, у яких є діти із спадковими хворобами, переважно уникають повторних вагітностей, що мотивується побоюванням мати ще одну хвору дитину. Рівень сучасної науки дозволяє проводити ранню дородову діагностику та попереджувати повторне народження хворої дитини.
У всьому світі преконцепційна профілактика та інвазивна пренатальна діагностика спадкових хвороб на ранніх термінах вагітності є для таких сімей шансом народити здорову дитину. Наявність здорової дитини у сім‘ях, де є хворі діти, є дуже позитивною із різних точок зору:
- в особі рідних братів та сестер хворі діти мають ширше коло спілкування, що дозволить їм краще адаптуватися у суспільстві;
- наявність здорової дитини наче «соціально реабілітує» родину та допомагає уникнути розлучень, що часто трапляються у таких сім‘ях, і таким чином, покращує сімейні умови для хворої дитини.
Що таке інвазивна пренатальна діагностика?
Мутації гена, що ведуть до спадкових хвороб, можуть бути ідентифікованими пренатально у плода (на ранніх етапах вагітності). У такому випадку матеріалом для дослідження є ДНК плода, виділена з клітин хоріона або навколоплідних вод. Забір матеріалу для пренатальної діагностики проводиться за допомогою інвазивних методів, таких як біопсія хоріону або плаценти, амніоцентез (забір навколоплідних вод) чи кордоцентез (забір крові плода). Забір матеріалу проводиться у гінекологічних стаціонарах і його терміни (терміни вагітності, на яких є найбільш безпечним проведення амніо- чи плацентоцентезу) залежить від наявних технічних можливостей та кваліфікації акушер-гінекологів. Матеріал для генетичного аналізу пересилається у лабораторні центри, де проводиться власне аналіз ДНК.
Низька інформованість пацієнтів про можливості пренатальної генетичної діагностики спадкових хвороби є перешкодою для її широкого застосування. Важливим є повідомлення про інформативну суть даного дослідження, що полягає лише у наданні інформації щодо стану плоду, а рішення щодо пролонгування чи переривання вагітності приймають батьки.
Результати пренатальних молекулярно-генетичних досліджень моногенних захворювань
Кожна родина, у якій були випадки народження дітей з аутосомно-рецесивними захворюваннями, має ризик повторного народження хворої дитини 25%. У результаті проведених ДНК-досліджень виявлено 384 подружні пари, які мають 25% ризик народження дитини з важким аутосомно-рецесивним захворюванням. Здійснено пренатальні молекулярно-генетичні дослідження мутацій генів ТРБМ (муковісцидоз), SMN (спінальна аміотрофія), NBN (синдром Ніймеген), ФАГ (фенілкетонурія) у сім’ях, де зареєстровані випадки цих аутосомно-рецесивних захворювань.
Усім сім’ям з аутосомно-рецесивними захворюваннями проведено медико-генетичне консультування та повідомлено їх про інформативність інвазивної пренатальної діагностики, яка є методом профілактики моногенних хвороб.
Інвазивна пренатальна діагностика проведена у 75 вагітних із подальшим використанням молекулярно-генетичних методів аналізу ДНК плода.
Фетальний матеріал для молекулярно-генетичної діагностики отримували методом трансабдомінальної аспіраційної біопсії ворсин хоріона у терміні 10–11 тижнів вагітності чи трансабдомінального амніоцентезу (АМЦ) у терміні 16–20 тижнів вагітності. Забір матеріалу здійснювали за допомогою голки з мандреном діаметром 20G, довжиною 205 мм під контролем ультразвукового сканера в умовах операційної. Протипоказом для проведення вважалася загроза переривання вагітності. Проводили виділення та очищення ДНК із клітин амніотичних вод чи ворсин хоріона.
Після проходження медико-генетичного консультування більшість жінок надала перевагу проведенню у них біопсії ворсин хоріона, проте, проведення інвазійної пренатальної діагностики у І–му триместрі вагітності було можливим у 18 із 41 випадків (44%).
За медико-генетичним консультуванням при плануванні вагітності звернулося 45 із 230 родин, де у пробанда верифіковано МВ (муковісцидоз). Серед них у 36 випадках хворий пробанд був єдиною дитиною. Із сорока п’яти родин 34 родини (75%) були повністю інформативними для проведення пренатальної молекулярно-генетичної діагностики мутацій гена ТРБМ, 9 – напівінформативними, 2 – неінформативними. Після роз’яснення процедури інвазійної пренатальної діагностики, на її проведення дало згоду 23 родини (10%). Серед сімей, які відмовилися від інвазивної пренатальної діагностики МВ, у п’яти родинах народилося семеро дітей. Серед них – три здорових гетерозиготних носії мутацій гена ТРБМ та чотири хворих на МВ. У 22 сім’ях здорових гетерозиготних носіїв гена ТРБМ під час вагітності проведено забір плодового матеріалу та подальше молекулярно-генетичне обстеження мутацій гена ТРБМ. В одній із родин пренатальна діагностика проводилася під час двох вагітностей. Результати ДНК-аналізу плода, терміни гестації на час проведення інвазивної пренатальної діагностики та методи наведені у таблиці 1.
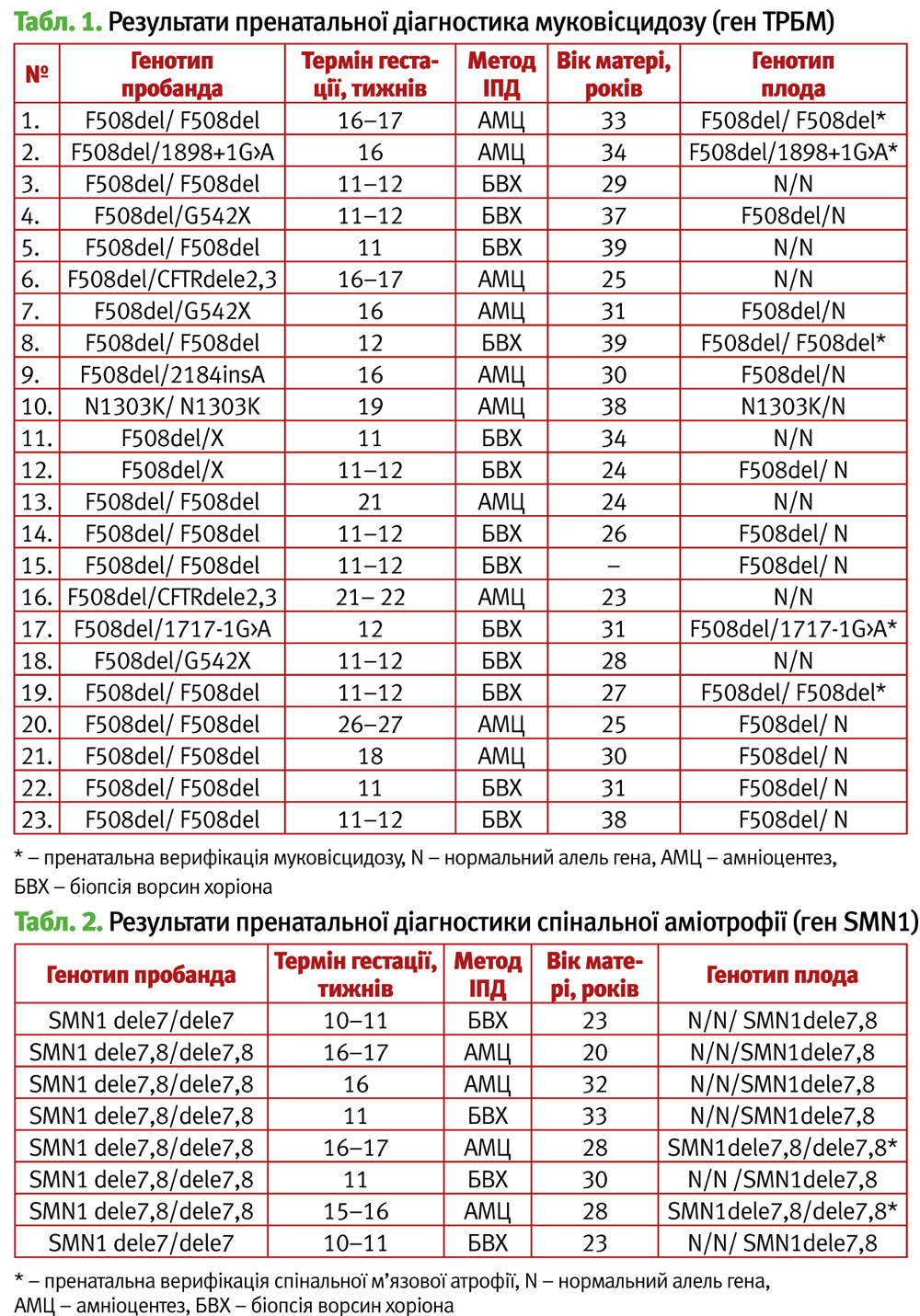
Аналізуючи результати 23 випадків пренатальної діагностики муковісцидозу, виявлено, що у 5 (22%) плодів діагностовано МВ. У 18 випадках у плодів виявлено відсутність мутацій гена ТРБМ у гомо – (30%) чи гетерозиготному (48%) станах.
Проведено медико-генетичне консультування та подальше генетичне тестування у 27 сім’ях, у яких один із партнерів був гетерозиготним носієм мутацій гена ТРБМ (серед них 9 – батьки пробандів у повторних шлюбах). У жодному із досліджених випадків ми не виявили мутацій гена ТРБМ у здорових партнерів. В усіх випадках у цих шлюбах народжені діти, у яких не зареєстровано клінічних проявів муковісцидозу.
Слід зазначити, що раніше, коли методи ДНК діагностики не були розвинуті, побутувала думка про те, що у випадку аутосомно-рецесивних захворювань зміна партнера є надійним методом запобігання повторного народження хворих дітей. Проте, слід відзначити, що нами зареєстровано два випадки повторного народження дітей, хворих на муковісцидоз, від наступних шлюбів. Таким чином, зважаючи на виявлену високу частоту гетерозиготного носійства мутацій гена ТРБМ в популяції, при плануванні вагітності носіями муковісцидозу обов’язковим є проведення аналізу ДНК у партнера.
Враховуючи встановлену нами частоту гетерозиготного носійства мутацій гена ТРБМ в Україні 1:29, ризик народження дитини, хворої на МВ, у шлюбі, де один із партнерів є гетерозиготним носієм гена ТРБМ, становить 1:116 (1:3364 – загально-популяційний ризик). У випадку негативного результату генетичного тестування у партнера (відсутність найбільш поширених мутацій гена ТРБМ) ризик народження дитини, хворої на МВ, знижується у п’ять разів і становить для жителів України 1:580.
Чоловіки, хворі на муковісцидоз, мають обструктивну азооспермію і у 95% випадків неплідні. У осіб жіночої статі, хворих на муковісцидоз, збережена нормальна репродуктивна функція, а основною проблемою є стан соматичного здоров’я жінки при вагітності та пологах. У трьох пробандів жіночої статі, у яких діагностовано МВ, народилися здорові діти. Народження дітей вагітними, хворими на муковісцидоз, є показником значного прогресу у лікуванні цього захворювання в Україні. Слід зазначити, що при відсутності мутацій гена ТРБМ у партнера пробанда не було показів для проведення інвазивної пренатальної діагностики, оскільки ризик народження дитини, хворої на муковісцидоз, у такого подружжя є низьким.
Для медико-генетичного консультування з метою планування вагітності звернулося 15 родин, у яких були випадки народження дітей, хворих на спінальну м’язову атрофію (СМА) і серед них у трьох родинах – по двоє хворих дітей. Після роз’яснення процедури інвазивної пренатальної діагностики, на її проведення під час вагітності дало згоду 7 родин із 61 (11,5%). Результати дородових досліджень спінальної аміотрофії наведені у Таблиці 2.
Із восьми проведених досліджень у двох випадках пренатально діагностовано спінальну м’язову атрофію. Серед сімей, які відмовилися від інвазивної пренатальної діагностики, у двох родинах народилося троє дітей – із них двоє хворих, які померли у перші шість місяців життя. Встановлено, що родини, де зареєстровано випадки спінальної аміотрофії, частіше зверталися для проведення пренатальної діагностики, ніж при інших досліджуваних нозологіях, що можна пояснити важкістю та інкурабельністю захворювання, високим рівнем летальності у ранньому віці.
Проте, слід відмітити, що нами зареєстровано унікальний випадок вагітності у 36-річної жінки, хворої на спінальну аміотрофію з генотипом SMN1dele7/dele7. Зважаючи на відсутність мутацій гена SMN1 у партнера, інвазивна пренатальна діагностика у цьому випадку не проводилася і у пари народилася здорова дівчинка. Такі дані свідчать про значну варіабельність клінічного перебігу спінальної м’язової атрофії в осіб з однаковими генотипами щодо гена SMN1.
Для медико-генетичного консультування з метою планування вагітності звернулося 10 із 34 родин, у яких були випадки народження хворих дітей із рідкісним аутосомно-рецесивним захворюванням – синдромом Ніймеген (важкий вроджений комбінований імунодефіцит, дизморфічні риси обличчя, мікроцефалія зі збереженим інтелектом). Серед них у трьох родинах – по двоє дітей із синдромом Ніймеген. Після роз’яснення процедури інвазивної пренатальної діагностики, на її проведення під час вагітності дало згоду 4 родини із 34 (11,7%). У сім’ях гетерозиготних носіїв мутації 657del5 гена NBN пренатальну діагностику проведено у шести випадках (Табл. 3).
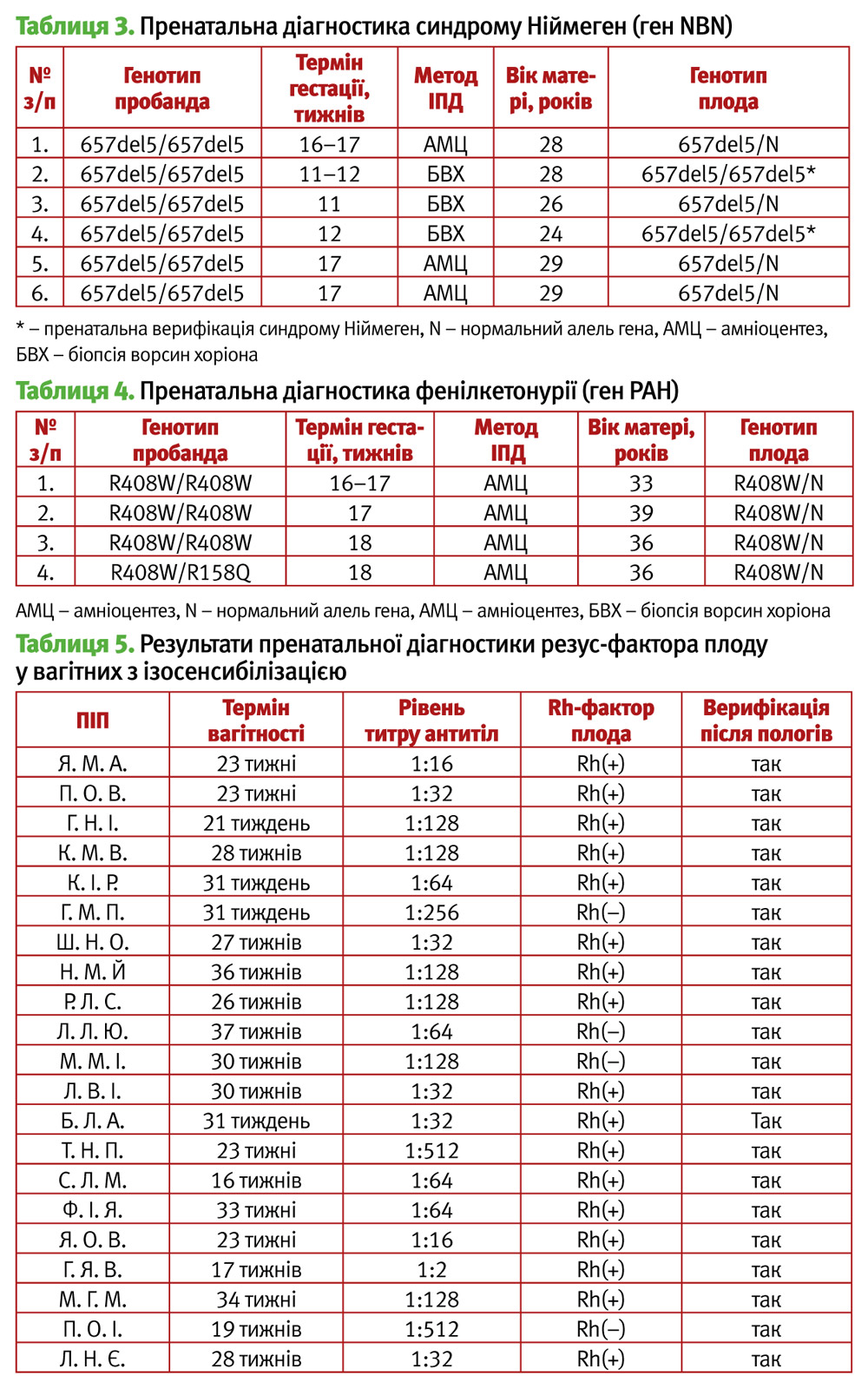
У результаті проведеного молекулярно-генетичного дослідження, у двох плодів встановлено наявність мутації 657del5 гена NBN у гомозиготному та у трьох – у гетерозиготному стані. Слід відмітити, що із шести випадків пренатальної діагностики синдрому Ніймеген, три проводилися в одній родині. У цій родині синдром Ніймеген був виявленим у двох старших дітей. За допомогою пренатальної діагностики під час третьої вагітності виявлено наявність мутацій гена NBN у гомозиготному стані, що свідчить про наявність синдрому Ніймеген у плода. Ця вагітність закінчилася самовільним перериванням у терміні 16 тижнів. У подальшому під час четвертої та п’ятої вагітностей проведено амніоцентез та виявлено мутацію 657del5 гена NBN у гетерозиготному стані. В обох випадках народилися здорові діти, які є гетерозиготними носіями синдрому Ніймеген.
Для медико-генетичного консультування з метою планування вагітності звернулося 20 із 61 родини, у яких проведено молекулярно-генетичну діагностику мутацій гена фенілаланінгідроксилази (ФАГ). Незважаючи на те, що фенілкетонурія є захворюванням, яке успішно піддається корекції, 4 із 64 родин (6,25%), у яких проведено молекулярно-генетичну діагностику мутацій гена фенілаланінгідроксилази (ФАГ), звернулися для проведення інвазивної пренатальної діагностики ФКУ (Табл. 4). Амніоцентез проведено у чотирьох матерів пробандів гомозигот за мутацією R408W та у плодів встановлено гетерозиготне носійство мутації R408W.
Усі родини, де діагностовано випадки моногенних захворювань, повідомлені про високий генетичний ризик повторного народження хворої дитини. Незважаючи на значний прогрес у терапії муковісцидозу, фенілкетонурії, синдрому Ніймеген, значному продовженні тривалості та покращенні якості життя таких пацієнтів, лікування вимагає від родини значних матеріальних, моральних та фізичних затрат. Батьки дітей, у яких верифіковано генетично детерміновані захворювання, в основному уникають повторних вагітностей, що мотивується боязню мати ще одну хвору дитину та погіршенням можливостей забезпечити їй необхідне лікування та догляд. Так, менше третини сімей запланували повторну вагітність після народження хворого пробанда, і лише 11–17% з них звернулися для проведення дородової діагностики. У чверті обстежених сімей хворий пробанд є єдиною дитиною у подружньої пари, що має негативні як біологічні (збільшення генетичного тягаря) так і соціальні наслідки (погіршення демографічних показників). Можливість визначення статусу плоду шляхом інвазивної пренатальну діагностики спонукає пари до планування вагітності та народження дітей.
Інвазивна пренатальна діагностика з елімінацією уражених плодів діє на зниження генетичного тягаря, проте така маніпуляція має цілу низку нерозв’язаних етичних проблем. Тому важливим є повідомлення пацієнтів про інформативну суть дослідження, що полягає лише у наданні інформації щодо стану плоду, а рішення щодо пролонгування вагітності приймають батьки. У ході проведення роботи нами виявлено низьку поінформованість пацієнтів про можливості пре- та постнатальної молекулярно-генетичної діагностики, що є перешкодою для ширшого запровадження цього аналізу. Усі вагітності із уражених плодом були перервані за бажанням батьків та медичними показами. У 33 випадках народилися здорові діти, у яких постнатально проведено генетичне тестування, результати якого співпали з даними пренатальної ДНК-діагностики. Окрім того, слід зазначити, що пари, у яких проводили пренатальну діагностику, при її відсутності уникали народження дітей взагалі.
Визначення резус-належності плода при підозрі резус-сенсибілізації
На даний час в світовій медичній практиці розроблені молекулярно-генетичні методи визначення резус-фактора, гетерозиготності за резус-фактором і груп крові у людини. У випадку, якщо резус-позитивні батьки є гетерозиготними за резус-фактором, то у резус-від’ємних матерів навіть за наявності обтяженого анамнезу плід у 50% випадків буде резус-від’ємним. Таким чином, для жінок з резус-конфліктною вагітністю з'явилася можливість проведення інвазивної пренатальної діагностики з метою визначення резус-фактора і групи крові за ДНК плода, виділеного з ворсин хоріона (10–15 тижнів вагітності) або з амніотичної рідини при проведенні амніоцентезу у термінах від 16 до 37 тижнів вагітності. Якщо результати молекулярно-генетичного аналізу показують, що плід резус-від’ємний, то немає необхідності у проведенні подальших інвазивних процедур, і такі пацієнтки виключаються з подальшого обстеження як групи ризику щодо розвитку гемолітичної хвороби плода.
Інвазивна пренатальна діагностика проведена за згодою у 26 жінок, у термінах від 16 до 37 тижнів вагітності. Із них 5 вагітним з резус-від’ємним типом крові резус-фактор плоду визначався при проведенні трансабдомінального амніоцентезу з метою каріотипування плода. Решта – 21 амніоцентез – проведено у вагітних, у яких неодноразово визначалися у крові антирезусні антитіла у титрі від 1:16 до 1:512, але за даними УЗ-діагностики, не виявлено ознак гемолітичної хвороби у плода. Результати пренатального визначення резус-належності плода подані у таблиці 5.
Як видно з отриманих результатів, у 4 вагітних із сенсибілізацією за резус-фактором, з високими титрами антитіл від 1:64 до 1:512 у плода, встановлений резус-від’ємний тип крові. Це дало можливість у 19,0% вагітних запобігти достроковому родорозрішенню та скоротити час перебування їх у стаціонарі. Вагітні з ізосенсибілізацією за резус-фактором, у яких плід був резус-позитивний, продовжили перебування у стаціонарі під ретельним спостереженням акушер-гінекологів. Отримані результати були верифікованні після народження дітей.
Визначення резус-фактора плоду пренатально дало можливість вирішити ряд проблем:
- Жінки, вагітні резус-від’ємним плодом, не хвилювалися про перебіг вагітності та пологів.
- У результаті встановлення резус-належності плоду вагітним не було необхідності регулярно визначати титр антитіл та проводити ультразвукове дослідження.
- Вагітним, у яких плід був резус-позитивним, проводилося ретельне спостереження: визначення титру антитіл, ультразвукове дослідження та кардіотахометрія. Були розширені покази до оперативного родорозв’язання.
Підсумки
Реєстрація усіх випадків гетерозиготного носійства аутосомно-рецесивних захворювань є ефективним заходом селективного скринінгу сімей високого ризику народження уражених дітей. У таких родинах слід провести медико-генетичне консультування та у випадку встановлення високого (25%) ризику – інвазивну пренатальну діагностику.
Інвазивна пренатальна діагностика з використанням аналізу ДНК залишається єдиним вірогідним способом встановлення статусу плоду щодо певного гена при кожній конкретній вагітності та ефективним методом профілактики нових випадків моногенних захворювань.
Інвазивна пренатальна діагностика має насамперед інформативну суть, і рішення щодо її проведення та подальшої тактики після отримання результатів ДНК аналізу належить батькам.
Пренатальне встановлення резус-належності плоду дозволяє зменшити кількість інвазивних втручань під час вагітності у резус-від’ємних матерів, вибрати оптимальний термін родорозрішення вагітних з резус-конфліктом, запобігти народження дітей з важкою формою гемолітичної хвороби та уникнути дотермінового родорозрішення у вагітних з резус-сенсибілізацією, у яких плід резус-від’ємний.
коментарів