

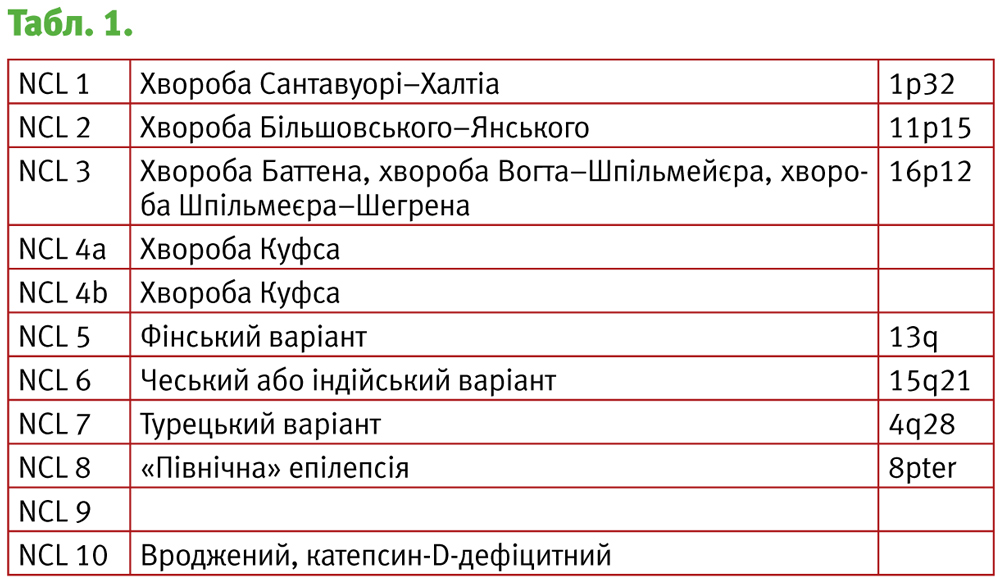
Під терміном «нейрональний цероїдний ліпофусциноз» (NCL) розуміють найбільш розповсюджену в дитячому віці групу клінічно та генетично гетерогенних нейродегенеративних захворювань, що характеризуються внутрішньоклітинним накопиченням автофлюоресцентних ліпопігментів у різних ультраструктурних локусах та клінічно проявляються втратою зору, швидко прогресуючою деменцією, епілептичними нападами, моторними розладами і ранньою смертю
У літературі захворювання даної групи описані як хвороба Баттена, хвороба Перрі, хвороба Шпільмейєра–Шегрена, хвороба Більшовського, хвороба Куфтса, хвороба Сантавуорі–Халтіа.
Перша згадка про нейрональний ліпоїдний ліпофусциноз з’явилась в 1826 році і належить Stengel, який описав перших 4 пацієнтів з Норвегії. Перший клініко-патологічний опис (1903) належить Batten. Він же охарактеризував NCL, як «сімейну нервово-м'язову дегенерацію». Також Batten був першим, хто диференціював NCL з хворобою Тея–Сакса у 1914 році. Приблизно в цей же час Vogt, Spielmeyer, Bielschwsky і Kufs також описали пацієнтів старшого віку з симптомами, схожими на описані Batten. Однак повноцінне вивчення захворювань даної групи почалося з 1980-х, з розвитком біохімії і молекулярної генетики.
Епідеміологія
Як вже було зазначено, дана група дуже гетерогенна за своїм клінічними, патофізіологічними та генетичними характеристиками, тому досить важко говорити про частоту даного захворювання. Однак, згідно з наявними даними, в США налічується близько 25000 сімей з встановленим діагнозом NCL.
Міжнародні дані: у фінській популяції NCL1 зустрічається з частотою 1:20 000, та 1 особа із 70 є носієм патологічного гену; поширеність NCL2 в світі — 0,6–0,7 на мільйон із захворюваністю на рівні 0,46 на 100 000 живих новонароджених; NCL3 – друга за частотою форма NCL в світі (7 випадків на 100 000 живих новонароджених в Ісландії). Найбільш висока поширеність NCL відзначається у скандинавських країнах, особливо в Фінляндії.
Патофізіологія
При всіх формах NCL відбувається накопичення у лізосомах клітин автофлюоресцентного ліпопігменту, який складається з білків сапозинів А і D і/або субодиниці з мітохондріальної АТФ-синтази.
Матеріал, що накопичується, характеризується автофлюоресценцією в зелено-жовтому спектрі при збудженні світлом довжиною від 340 до 360 нм.
При гістохімічному фарбуванні матеріал дає позитивну реакцію на кислу фосфатазу і забарвлюється суданом чорним В, що свідчить про присутність фосфоліпідів. Він також є ШИК-позитивним, що може вказувати на високий вміст вуглеводів. Більшість з цих властивостей ідентична з властивостями так званих пігментів «зношування», або «старіння», яких ще називають ліпофусцинами або цероїдами. При електронній мікроскопії нейрональних і екстранейрональних тканин виявляють цитосоми, заповнені агрегатами характерної морфології, що описуються, як «криволінійні» профілі, за типом «відбитків пальців», «прямолінійні» профілі, гранулярні осміофільні відкладення.
Ліпофусцин – дрібний, гранулярний, золотисто-коричневий пігмент, що складається з фосфоліпідів і білків. Ліпофусцин накопичується в організмі тварин і людини в нормі, у міру їх зростання і старіння. Залежно від віку, умов утворення і локалізації розрізняється за гістохімічними і ультраструктурними характеристиками, спектрами флуоресценції і поглинання, розчинності в органічних розчинниках.
Цероїд – ліпопігмент, який утворюється в макрофагах шляхом гетерофагії при резорбції ліпідів. Містить нерозчинні в спирті ремнанти окислених ЛПНЩ, які здатні витримувати лізосомальний гідроліз в макрофагах. Механізм утворення цероїду можна уявити наступним чином: після ендоцитозу ліпідів з утворенням ліпофагосом, відбувається їх часткове перетравлювання у вторинних лізосомах з утворенням третинних лізосом, або телолізосом, які містять речовину, що зветься цероїдом.
Спочатку класифікація NCL ґрунтувалася на віці початку маніфестації та симптоматиці захворювань і налічувала 4 типи:
- Тип I – рання дитяча форма.
- Тип II – пізня дитяча форма. Хвороба Більшовського–Янського.
- Тип III – підліткова форма. Хвороба Баттена–Шпільмеєра–Фогта.
- Тип IV – пізня форма. Хвороба Куфса.
На теперішній момент класифікація доповнена даними генетичних досліджень і набула наступного вигляду (табл. 1).
Всі форми захворювання мають аутосомно-рецесивний тип спадкування.
Характеристика окремих типів NCL
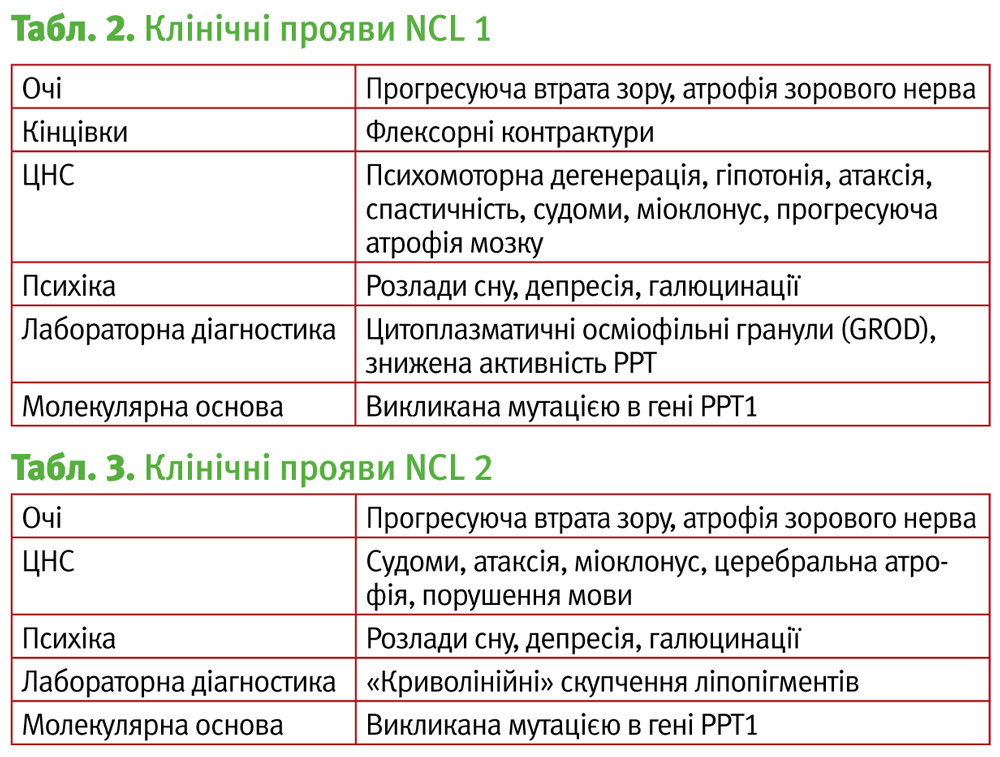
NCL 1, хвороба Сантавуорі–Халтіа, інфантильний ліпофусциноз.
Захворювання пов'язане з мутацією гена 1р32, відповідального за синтез пальмітоїл-протеїн тіроестерази-1 (PPT-1), яка видаляє ацильні групи цистеїну на ліпопротеїдах ЕПС.
Розрізняють наступні форми захворювання:
- Класична інфантильна
1968 – Herbert et al. описав прогресуючу енцефалопатію у дитини з фінської сім'ї.
1973 – Santavuori et Haltia описали основні клінічні та морфологічні особливості даної форми – мікроцефалія, гіпотонія, гіперзбудливість, когнітивні розлади, зорові порушення, атаксія, екстрапірамідні порушення, спастичність, міоклонус, втрата моторних і соціальних навичок до 2 років, смерть до 6–13 років.
- Пізня інфантильна
1979 – Becker в Німеччині описав дитину, у якого зорові і ментальні розлади почалися до 3 років, з попередніми міоклонічними нападами. Відмічалось прогресуюче зниження когнітивних функцій, клінічно форма нагадує NCL2, смерть до 10–13 років.
- Ювенільна
1995 – опис належить Philippart et al. і Hofman and Taschner. Спостерігалася втрата зору і здатності до придбання нових навичок до 5–7 років, клінічно схожа з NCL3, але з пізнім початком епілептичних нападів і більш ранніми моторними порушеннями
- Доросла форма
2001 – Van Diggelen описав 2 сестер, з початком NCL1 в дорослому віці і встановленими мутаціями гена РРТ1.
2007 – Ramadan et al. був описаний випадок NCL1 у 24-річної жінки. Виявлено маніфестацію до 30 років, психічні порушення з прогресуючою когнітивною дисфункцією, атаксію, паркінсонізм, атрофію зорового нерва; пацієнти з цією формою захворювання доживають до 50 років.
В цілому характеристика даного типу відображена в табл. 2.
NCL 2, хвороба Більшовського–Янського, пізній інфантильний NCL.
Мутація гена 11р15.5, відповідального за синтез лізосомльного ферменту трипептид-пептідази-1, що відщеплює трипептиди з N-поліпептидного кінця.
Розрізняють наступні форми:
- Пізня інфантильна
1926 – Hassin належать перші відомості про дану форму захворювання.
1957 – Seitelberger та ін. визначають в літературі 28 випадків. Маніфестація у віці 2–4 років, епілептичні напади, зниження когнітивних функцій, атаксія, міоклонус, екстрапірамідні розлади, повна втрата зору до 4–6 років, смерть у другій декаді життя або раніше.
- Ювенільна
1977 – Дана форма була вперше виділена Andermann та ін. Йому ж належить опис 17 випадків захворювання. Маніфестація у 6–8 років, прогресуюче зниження когнітивної функції, судоми, атаксія, моторні порушення, різні порушення зору, деякі пацієнти доживають до 40 років.
Загальна характеристика подана у табл. 3.
NCL 3, хвороба Баттена, хвороба Вогта–Шпільмейєра, хвороба Шпільмейєра–Шагрена.
Мутація гена 16р12.1, що кодує білок, який складається з 438 амінокислот та є частиною мітохондріальної мембрани. Найбільш частим видом мутації даного гена є 1.02-kb делеція.
Розрізняють:
- Ювенільну форму
1904 – Баттен описав пацієнтів з даним захворюванням та визначив прогресуюче зниження зору з 4–7 років, що приводить до повної втрати зору у 10 років, дизартрію, зниження когнітивних функцій, епілептичні напади, психічні порушення у вигляді порушення соціальної адаптації та ідентифікації, зниження уваги, підвищену агресію, паркінсонізм, міоклонус, розлади сну, мозочкові розлади та розлади екстрапірамідної системи.
Загальна характеристика подана у табл. 4.
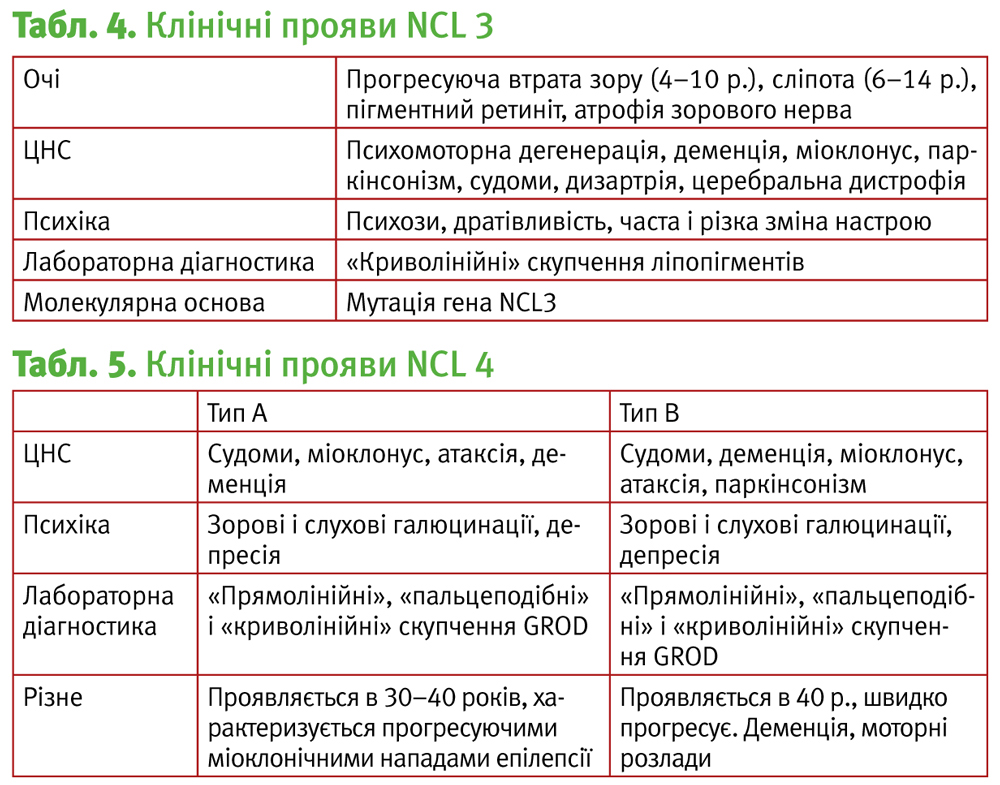
NCL 4, хвороба Куфса, NCL дорослих.
Мутація гена NCL4. Маніфестує даний тип, як правило, у віці 30 років, проте є дані, згідно з якими можлива маніфестація і у віці 11 років.
1925 – Куфс вперше описав випадок дорослого NCL, з проявом захворювання у 26 років і смертю пацієнта в 34 роки.
Виділяють два типи даного захворювання:
- тип А: відзначаються прогресуючі міоклонічні епілептичні напади, деменція, атаксія, пірамідні і екстрапірамідні розлади.
- тип В: деменція, моторні порушення, атаксія, маніфестація можлива і після 50 років (табл. 5).
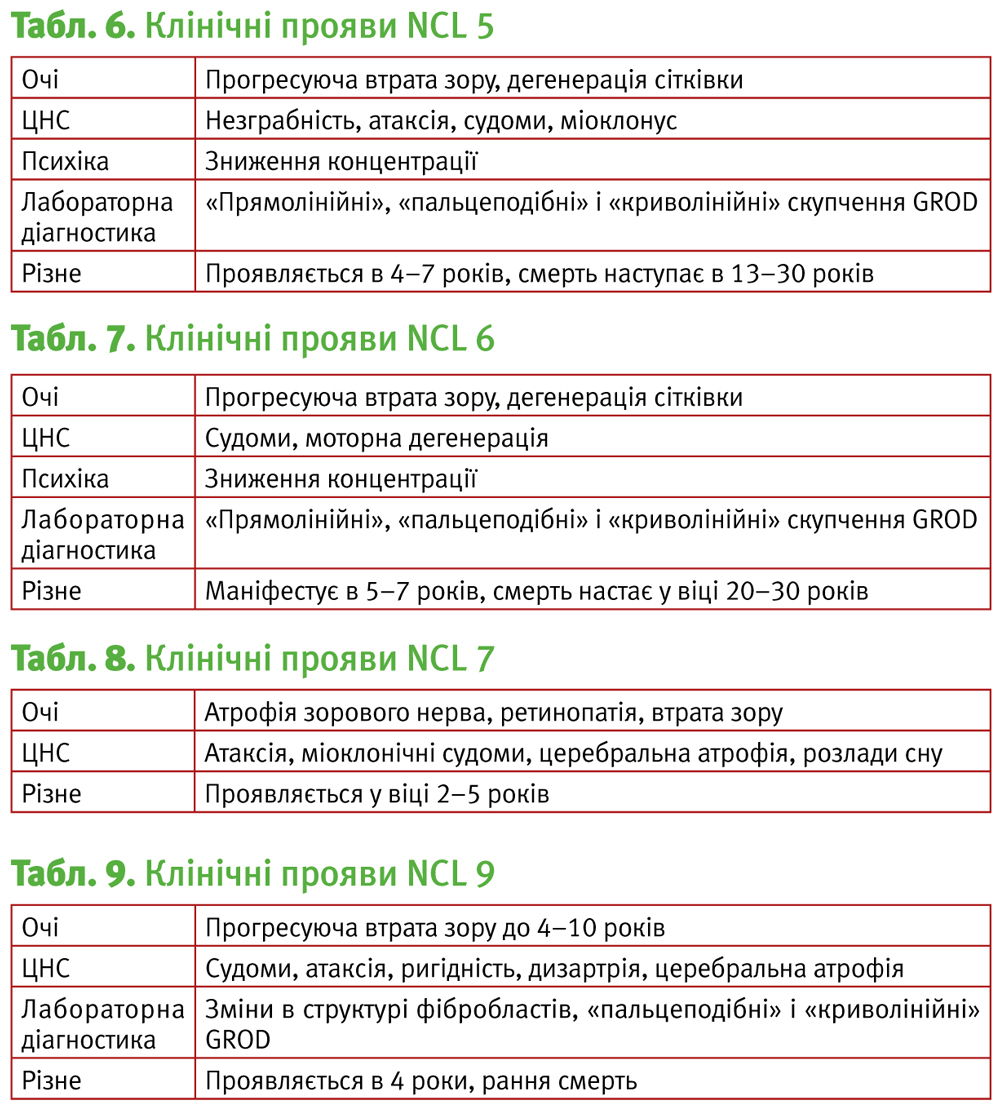
NCL 5, пізній інфантильний, фінський варіант.
Мутація гена 13q21.1-q32,що кодує трансмембранний білок, який складається з 407 амінокислот.
Перший опис хвороби даний Santavuori у 1982 р. Він повідомив про 18 фінських сімей, в яких були хворі на NCL. Маніфестація у віці 4–7 років, клінічно хвороба нагадує NCL2, але протікання більш повільне, смерть настає у другій-третій декаді життя.
Загальна характеристика подана у табл. 6.
NCL 6, пізня інфантильна форма, чеський або індійський варіант.
Мутація гена 15q21-q23, що кодує мембранний білок, який складається з 311 амінокислот.
1997 – Sharp et al. описують 2 сім'ї із захворюванням, що гістологічно подібне до NCL2. Прояви хвороби, як правило, починаються від 18 місяців до 8 років у вигляді зниження або втрати зору, судом, втрати моторних навичок до 4–10 років; смерть настає у другій-третій декаді життя. Варіант захворювання клінічно схожий з NCL2 (табл. 7).
NCL 7, турецький варіант.
Захворювання викликане мутацією в гені 4q28.1-q28.2, що кодує лізосомальний мембранний переносник.
1999 – Wheller et al. описує пацієнтів з так званим турецьким варіантом NCL.
2005 – Ranta вивчає 7 турецьких сімей з даним захворюванням. Захворювання характеризується атрофією зорового нерва, атаксією, міоклонічними судомами, церебральною і мозочковою атрофією, розладами сну (табл. 8).
NCL 8, пізній інфантильний варіант, «північна епілепсія».
Мутація гена 8pter-p22, що кодує трансмембранний білок, який складається з 286 амінокислот.
1999 р. – Wheeller повідомляє про виявлення 6 пацієнтів з відомим для NCL фенотипом, але не встановленими локусом мутації.
Для NCL8 визначено 5 різних мутацій, але місценс-мутація (R24G), призводить до прогресуючої епілепсії з затримкою розумового розвитку (PERM).
Відповідно розрізняють:
- турецький варіант пізнього інфантильного NCL – проявляється у 3–7 років, прогресуюча втрата зору, судоми, інтелектуальні порушення, дизартрія, міоклонус, атаксія.
- «північну» епілепсію – епілептичні напади у 5–10 років, повільне прогресуюче зниження розумового розвитку, можливе зниження зору, деякі пацієнти доживають до 60 років.
NCL 9.
Даний тип описаний у 2004 році Schulz на 2 сестрах з Сербії та братах з Німеччини. У сестер відзначалося зниження зору, прогресуюча атаксія і судоми у 4 роки. До 10 років була втрачена здатність до самостійної ходьби. Подібна симптоматика спостерігалася і у двох братів. Зниження когнітивних функцій почалося з 6 років, атаксія і ригідність відзначалися з 9 років. Молодший із братів помер у 15 років від пневмонії,а старший, який страждав згодом дисфагією і галюцинаціями, помер у віці 19 років. Імовірно, що ген, мутація якого викликає захворювання, кодує білок, що бере участь у регуляції активності дигідроцерамідсинтетази (табл. 9).
NCL 10. Вроджений, катепсин-D-дефіцитний.
Мутація гена 11p15.5, що кодує катепсин D. Перші згадки відносяться до 1941 р. Norman та Wood, а також до 1954 р. Brown. Дана форма описана в 2006 році Steinfeld у групі із 25 дітей з не ідентифікованим NCL-подібним захворюванням. Характеризується наступними ознаками: мікроцефалія, прогресуюча втрата зору, епілептичні напади, епілептичний статус, атаксія, спастичність, ригідність, церебральна атрофія.
Діагностика
У діагностиці NCL використовують дані електрофізіологічних та гістологічних досліджень, а також різних методів візуалізації.
Магнітно-резонансна томографія (МРТ): NCL1 – слабка церебральна атрофія, прогресуюча після 4 років, зниження T2-сигналу в області таламуса, прогресуюче зниження контурованості білої речовини на T2, атрофія мозочка з 3 років, витончення поясної звивини. CLN2 – прогресуюча, особливо субтенторіальна, атрофія мозку. CLN3 – церебральна атрофія, зазвичай після 15 років. NCL6 – поширена церебральна і мозочкова атрофія.
Позитронно-емісійна томографія (ПЕТ): NCL2 – генералізований гіпометаболізм. CLN3 – гіпометаболізм в області шпорної борозни.
Магнітно-резонансна спектроскопія (МРС): NCL1 – майже повна втрата N-ацетиласпартату, редукція креатин- і холін-вмісних сполук, зростання міоінозитолу, зростання вмісту лактату в сірій і білій речовині.
ЕЕГ: NCL1 – втрата атенуації при відкриванні очей, згладжування «веретен сну». NCL2 – потиличні спайки 1-2 Hz зі світловим подразником.NCL3 – спайки і повільно-хвильові комплекси.
Електроретинограма: NCL1 (інфантильна форма) – неможливо записати до 3 років. NCL1 (ювенільна форма) – неможливо записати. NCL2 – неможливо інтерпретувати, в подальшому зникає. NCL3 – ранні аномалії при реєстрації.
Зорові викликані потенціали: NCL1 – неможливо записати з 4 років. NCL2 – відзначаються аномалії, але інтенсивність сигналу знижується на фінальних стадіях. NCL3 – ранні аномалії при реєстрації.
Гістологічне дослідження. NCL1 – майже повне зникнення кортикальних нейронів. NCL3 – вакуолізовані лімфоцити, селективний некроз клітин-сателітів 2 і 3 шару кори, і втрата пірамідних клітин 5 шару. NCL5 – дегенерація нейронів неокортексу і мозочка, дегенерація 3 і 5 шарів кори, багатоядерні нейрони в 3 шарі кори. NCL6 – втрата нейронів 5 шару, зниження кількості клітин Пуркіньє, відсутність SCMAS в печінці, наднирковій і підшлунковій залозах. NCL8 – дегенерація клітин 5 шару і нейронів гіпокампу, зниження SCMAS в клітинах Пуркіньє.
Остаточний діагноз верифікується за допомогою ДНК-тестування з визначенням мутантного гена.
Диференціальна діагностика
Дифдіагностика проводиться з абсансною епілепсією, комплексними парціальними судомами, допамін-чутливою дистонією, Epilepsia Partialis Continua, атаксією Фрідрейха, хворобою Халлевордена–Шпатца, хворобою Гантінгтона, Retinitis pigmentosa, синдромом Ретта, метаболічними та інфекційними ушкодженнями ЦНС.
Лікування
Специфічної патогенетичної терапії не розроблено. Лікування симптоматичне (антиконвульсанти), використання антиоксидантів безрезультатні, проводилися спроби пересадки кісткового мозку, але ця методика не виявилася дієвою.
У якості ілюстрації наводимо власне спостереження випадку нейронального цероїдного ліпофусцинозу ІІ типу у дитини.
Хлопчик Д., 5 років, зі скаргами на напади судом, які почастішали у динаміці.
Анамнез: друга вагітність, перші пологи. Перша вагітність закінчилась викиднем. Пологи – кесарів розтин на 40-му тижні, вага при народженні 4050 г. Постнатальний період в нормі. Почав стояти у 7–8 місяців, ходив самостійно в 10 місяців. Мовні навички: перші слова у віці одного року.
В 3 роки у дитини почалися судомні напади різного типу, включаючи генералізовані міоклонічні судоми. Хода дитини стала нестабільною. Спостерігалося легке відставання в мовному розвитку. Лікувався у міському неврологічному відділенні. Було встановлено діагноз: Злоякісна міоклонічна епілепсія. Аутизм. Призначено лікування комбінацією антиконвульсантів, яке не принесло бажаного ефекту. З січня 2015 р. (4 роки) хлопчик не вимовляє слів, поведінка аутична. Спостерігалась мовна регресія протягом 9 місяців. В останні 6 місяців спостерігався моторний і когнітивний регрес. З квітня 2015 р. – посилення судом і регресія, в основному, мовних навичок. Моторно — наросла м’язова слабкість, іноді міг йти сам, іноді потребував допомоги. Також відмічалася регресія контролю над природними відправленнями.
Хлопчик був спрямований на обстеження у Центр орфанних захворювань, де встановлено діагноз: Епісиндром. Затримка мовного розвитку. Було рекомендовано консультування і обстеження в медичному центрі «Асафа-Рофе» (Ізраїль) на предмет діагностики спадкових нейродегенеративних захворювань.
У квітні 2015 р. в медичному центрі «Асафа-Рофе» були проведені наступні дослідження:
Лабораторні аналізи: електроліти та функції печінки в нормі, функції нирок у нормі, СРБ нульовий. Вальпроєва кислота 34,4. ОАК: лейкоцити 8,6 гемоглобін 11,7, тромбоцити в нормі.
Метаболічні дослідження: вміст у крові амінокислот, карнітину, ацилкарнітинів, аміаку, піровиноградної кислоти – без відхилень від норми. Рівень лактату трохи підвищений — 25 (норма до 18), сеча на сечову кислоту — в нормі.
ЕЕГ: полівогнищева епілептична активність (ліва лобова частка, права тім'яна частка). Короткі (1–5 секунд) спалахи генералізованих тета-хвиль високої напруги зі змішаними піками.
МРТ мозку: гіпоплазія мозочка, зменшення об'єму супратенторіальної та інфратенторіальної тканини мозку, генералізована атрофія паренхіми, дилатація шлуночків.
Генетична діагностика: проведено генетичний чіп – без хромосомних змін, пов'язаних з високою ймовірністю з відомим генетичним або неврологічним синдромом.
Секвенування екзома: виявлено в екзоні 6 гетерозиготний патогенний варіант гену ТТР-1, с.622С>Т, також у екзоні 7 гетерозиготний варіант гену ТТР-1, с.833А>С, що свідчить про підтвердження діагнозу «Нейрональний цероїдний ліпофусциноз, тип 2».
Клінічний діагноз: Нейрональний цероїдний ліпофусциноз, тип 2. Генералізована епілепсія з міоклонічним компонентом.
Рекомендовано:
- Почати терапію фелбаматом в дозі 150 мг двічі на день;
- Почати терапію клобазамом в дозі 10 мг двічі на день;
- ЕЕГ через три місяці;
- Контрольна МРТ через рік для спостереження;
Призначена терапія не принесла бажаного результату. Періодично виникали судоми. З вересня 2016 року, судоми відзначались покілька разів на тиждень. Протягом останніх трьох днів перед госпіталізацією було 3–5 судом в день. 21.09.16 стан погіршився, додому була викликана бригада швидкої допомоги, вдома введено сибазон. Дитину з епістатусом було госпіталізовано у ВАІТ ДМКЛ №6.
При огляді у ВАІТ: стан дитини тяжкий, обумовлений неврологічною симптоматикою. t – 36,5ОС; ЧСС – 90/хв.; ЧД – 26/хв.; АТ – 110/60 мм рт. ст.; SpO2 – 99%. Свідомість – медикаментозний сон. Менінгеальні симптоми негативні. Зіниці D=S, обличчя симетричне. Плаваючі рухи очних яблук. Короткочасні тонічні напруження кінцівок. Шкірні покриви бліді, теплі, висипань не спостерігається. Тургор м’яких тканин та еластичність шкіри достатні. Видимі слизові порожнини рота рожеві, помірної вологості. Задишки та видимих гіпоксичних розладів при диханні атмосферним повітрям немає.
Аускультативно: в легенях дихання жорстке, проводиться рівномірно в усі відділи. Перкуторно над легенями визначається легеневий звук з обох боків. Тони серця приглушені, ритмічні. Видимих набряків немає. Живіт м’який, доступний глибокій пальпації. Печінка +1,0 см. Біля краю реберної дуги. Селезінка не пальпується. Фізіологічних відправлень при огляді не було.
Лабораторні дослідження: загальний аналіз крові, загальний аналіз сечі, нирково-печінковий комплекс, електроліти, глюкоза – у межах вікової норми.
ЕЕГ: полівогнищева епілептична активність.
Консультація невролога: нейрональний цероїдний ліпофусциноз. Симптоматична епілепсія. Груба затримка психо-моторного розвитку. Стан після епілептичного нападу.
Клінічний діагноз: Нейрональний цероїдний ліпофусциноз, ІІ тип. Симптоматична епілепсія. Епістатус.
У відділенні реанімації отримував наступну терапію: препарати вальпроєвої кислоти, карбамазепін, клобазам. Сибазон за показаннями.
Через 3 дні стан дитини покращився. Судомний синдром був купіруваний. Дитина у стабільному стані переведена для подальшого лікування у відділенні неврології.
Висновки
Сьогодні перелік орфанних захворювань стає все об'ємнішим, а можливості їх діагностики – доступнішими. У даному випадку наявність у дитини поєднання епілептичних нападів із ознаками нейродегенеративного захворювання стало підставою для пошуку генетичної патології. Уточнення діагнозу дозволяє визначити прогноз та в ряді випадків оптимізувати терапію. Тому ми вважаємо за доцільне нагадати основні показання для спрямування пацієнта з неврологічними порушеннями у медико-генетичні центри та центри метаболічних захворювань:
- епілептичні судоми, що важко піддаються належній медикаментозній корекції;
- ознаки нейродегенеративних захворювань центральної нервової системи у поєднанні з захворюваннями органів зору, слуху, атаксіями та моторними порушеннями;
- МРТ-ознаки лейкодистрофії нез’ясованої етіології;
- невпинна втрата психомоторних навичок, набутих раніше;
- моторні порушення у поєднанні з мальформаціями Денді–Уокера та іншими гіпоплазіями мозочка;
- міопатичні синдроми;
- полінейропатії, не пов’язані з інфекційно-алергічною природою;
- розумова відсталість, аутична поведінка та інші порушення поведінки, поєднані з набутими порушеннями зору, слуху та моторними порушеннями.
коментарів